




Diagnosis of neurofibromatosis 1 (NF1) diagnosis is challenging in young children without a family history of NF1. The aims of this study were to estimate diagnostic delays in children without a family history of NF1 and to examine the effects of considering café-au-lait macules and skinfold freckling as a single diagnostic criterion.
Patients and methodsRetrospective, descriptive, observational study of all patients diagnosed with NF1 before the age of 18 years who were seen at our hospital. The medical records of those included were reviewed to identify the date on which the diagnostic criteria of NF1 were objectified. The patients were categorized into 2 groups: those with a known parental history of NF1 and those without. Café-au-lait macules and skinfold freckling were assessed as a single diagnostic criterion, and genetic evidence was considered to confirm highly suspicious cases.
ResultsWe studied 108 patients younger than the age of 18 years with a diagnosis of NF1. Mean (SD) age at diagnosis was 3.94 (±3.8) years for the overall group, 1 year for patients with a parental history of NF1, and 4 years and 8 months for those without. Diagnosis was therefore delayed by 3 years and 8 months in patients without a family history.
ConclusionSkin lesions were the first clinical manifestation of NF1 in most patients. We believe that the National Institutes of Health's diagnostic criteria for NF1 should be updated to aid diagnosis in young children.
El diagnóstico de la neurofibromatosis 1 (NF1) plantea dificultades en niños sin antecedentes familiares durante la primera infancia. En este estudio pretendemos estimar la demora diagnóstica de los pacientes sin antecedentes familiares de NF1 y definir la repercusión de considerar las manchas café con leche y las efélides como un único criterio diagnóstico.
Pacientes y métodosEstudio observacional descriptivo retrospectivo en el que se revisaron los hitos diagnósticos de la NF1 en las historias clínicas de los pacientes menores de 18 años atendidos en nuestro centro. Distribuimos a los pacientes en dos grupos en función de la existencia de antecedentes de NF1 entre sus progenitores, considerando las manchas café con leche y las efélides como un único criterio y aceptando el estudio genético como criterio de confirmación en casos de elevada sospecha.
ResultadosSe incluyeron en el estudio 108 menores con diagnóstico de NF1. La edad media de diagnóstico en nuestra serie fue de 3,94 años (desviación estándar: ±3,8 años). En el grupo 1, sin antecedentes, la edad media de diagnóstico fue de 4 años y 8 meses, mientras que en el grupo 2, con antecedentes, fue de 12 meses, siendo la demora en el diagnóstico de 3 años y 8 meses entre ambos grupos.
ConclusiónLas lesiones cutáneas representan, en la mayoría de los casos, las primeras manifestaciones clínicas de la enfermedad. Consideramos necesaria la actualización de los criterios diagnósticos del NIH con el fin de facilitar el diagnóstico en los primeros años de vida.
The diagnosis of neurofibromatosis type 1 (NF1) (#162200) is based on confirmation of at least 2 of the 7 diagnostic criteria established by the National Institutes of Health (NIH) in 1987.1 Several modifications to these original criteria were recently proposed by a group of experts2 (Table 1). An estimated 50% of NF1 diagnoses are made by dermatologists.3
National Institutes of Health (NIH) Diagnostic Criteria for Neurofibromatosis 1 (NF1) and Revised Criteria Proposed by Legius et al. in 2021.a
| NIH criteria | 1. Six or more café-au-lait macules (>5mm in prepubertal individuals and >15mm in postpubertal individuals)2. Axillary or inguinal freckling (Crowe's sign) or inguinal region.3. Two or more neurofibromas of any type or 1 plexiform neurofibroma4. Optic pathway glioma5. Two or more Lisch nodules in eye examination6. A distinctive osseous lesions (sphenoid dysplasia or thinning of long bone cortex with or without pseudarthrosis)7. First-degree relative with NF1 |
| Revised criteria proposed by Legius et al. | A. The diagnostic criteria for NF1 are met in an individual who does not have a parent diagnosed with NF1 if 2 or more of the following are present:1. Six or more café-au-lait macules (>5mm in prepubertal individuals and >15mm in postpubertal individuals)2. Axillary or inguinal freckling (Crowe's sign)b3. Two or more neurofibromas of any type or 1 plexiform neurofibroma4. Optic pathway glioma5. Two or more iris Lisch nodules identified by slit lamp examination or 2 or more choroidal abnormalities – defined as bright, patchy nodules imaged by optical coherence tomography/near-infrared reflectance imaging6. A distinctive osseous lesion such as sphenoid dysplasia,canterolateral bowing of the tibia, or pseudarthrosis of a long bone7. A heterozygous pathogenic NF1 variant with a variant allele fraction of 50% in apparently normal tissue such as white blood cellsB. A child of a parent who meets the diagnostic criteria specified in A merits a diagnosis of NF1 if 1 or more of the criteria in A are present. |
Although the diagnostic features of NF1 may be easy to recognize in adults,4 young children without a family history of this disease often have to wait several years before a definitive diagnosis can be made. Based on the largest pediatric series of NF1 published, the mean age at diagnosis lies between 2.65 and 4.5 years.3,5–7 In the absence of a family history of NF1, observation of multiple café-au-lait macules (CALMs) in the first months of life is nearly always the first step toward suspecting or establishing a diagnosis.
The aims of this study were to estimate diagnostic delays in children without a family history of NF1 and to examine the effects of considering café-au-lait macules (CALMs) and freckling as a single diagnostic criterion.
Material and MethodsRetrospective, descriptive, observational study in which we reviewed the medical records of all patients diagnosed with NF1 before the age of 18 years who were seen at the pediatric NF1 unit of our hospital between May 1, 2012 and April 30, 2016. Inclusion criteria were confirmation of 2 or more NIH criteria or, in patients with CALMs and freckling only, confirmation of 2 criteria and detection of mutations in the NF1 gene. Informed consent from the patients’ parents or guardians was also required.
The study protocol was approved by the ethics and research committee of Hospital Niño Jesús in Madrid (R-0024/16, No. 11/16, September 27, 2016).
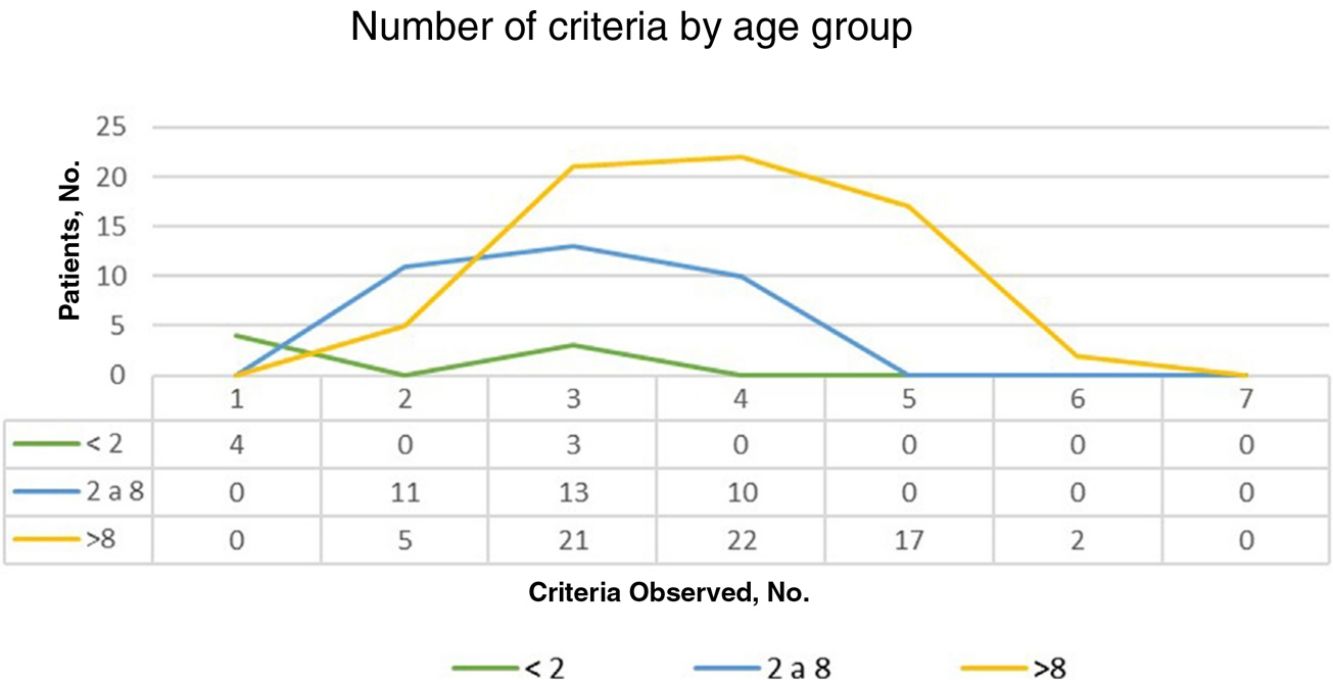
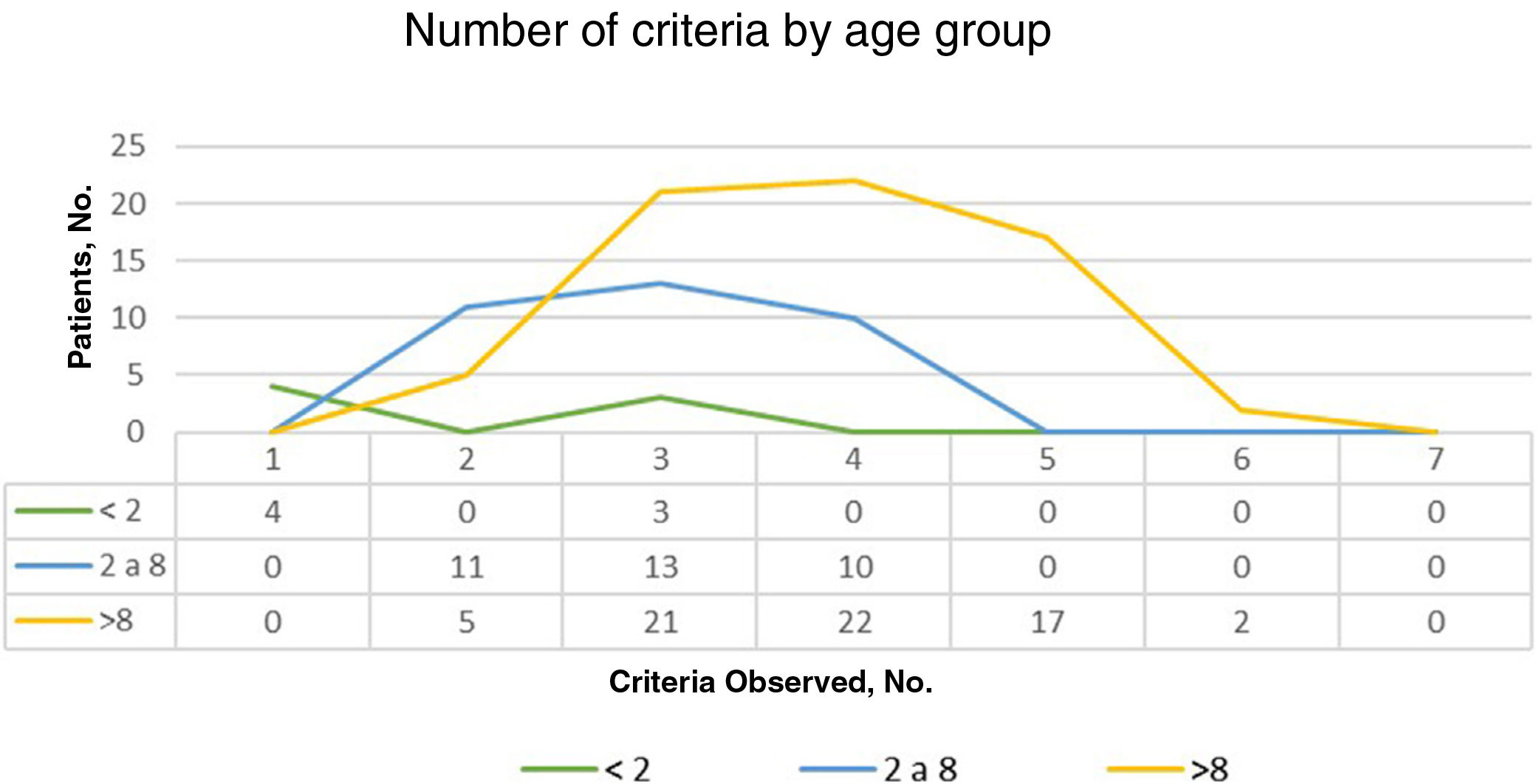
ResultsOf the 135 children with suspected NF1 seen during the study period, 108 met the inclusion criteria. At diagnosis, these patients had a mean (SD) age of 3.94 (3.8) years and a median age of 3. Patients were classified into 2 groups according to whether or not they had a family history of NF1. Group 1, comprising those without a known family history or with a de novo mutation, was formed by 86 patients (79.63%). The diagnostic criterion of 6 or more CALMs measuring more than 5mm in the greatest diameter was the first sign that led to a definitive or suspected diagnosis in all cases. The mean (SD) age in this group was 4.7 (3.87) years (4 years and 8 months) and the median age was 4 years. A definitive diagnosis was made in 3 of the patients on the day of the visit on discovering that 1 of the parents also had NF1 that had previously gone unrecognized. Genetic testing confirmed the diagnosis in 19 patients (17.59%) with pigmentary features only (Table 2). Group 2, comprising patients with a parental history of NF1, was formed by 22 patients (20.37%). The mean age at diagnosis in this group was 0.99 (1.31) years (12 months) and the median was 0.62 years (7.5 months). The rest of the characteristics are summarized in Table 2. Fig. 1 shows the number of criteria according to age group. The youngest age group (< 2 years) comprised patients who in addition to pigmentary features had an NF1 mutation confirmed by molecular analysis. Overall, most of the patients had 3 or 4 criteria, and none of them had all 7 (Fig. 1).
Distribution of National Institutes of Health (NIH) Diagnostic Criteria for Neurofibromatosis 1 by Mean and Median Ages at Diagnosis.
| Suggestive criteria | Definitive criteria | No., % | Mean | Median | SD | |
|---|---|---|---|---|---|---|
| Group 1n=86 (79.63%) | CALMs±freckling | Diagnosis of NF1 in one of the parents on the day of the child's visit | 3, 2.79% | 7.08 | 10 | 5.94 |
| Mean (SD): 4 y and 8 mo (3.87 y) | Neurofibromas | 23, 21.39% | 5.71 | 4 | 5.16 | |
| Optic pathway glioma | 16, 14.81% | 3.38 | 3 | 1.68 | ||
| Lisch nodules | 24, 22.32% | 5.94 | 6 | 3.51 | ||
| Osseous dysplasia | 1, 0.93% | 1.5 | 1.5 | NA | ||
| Genetic study | 19, 17.59% | 2.96 | 2.5 | 2.73 | ||
| Group 2n=22 (20.37%) | First-degree relative with NF1 | CALMs | 19, 17.59% | 0.88 | 0.5 | 1.28 |
| Mean (SD): 12 mo (1.31 mo) | Freckling | 1, 0.93% | 0.5 | 0.5 | NA | |
| Neurofibroma | 1, 0.93% | 3.5 | 3.5 | NA | ||
| Genetic study | 1, 0.93% | 1 | 1 | NA | ||
Abbreviations: CALMs, café-au-lait macules; NA, not assessable.
During the study period, and prior to the definitive diagnosis, nevus anemicus was observed in 22 patients and juvenile xanthogranuloma in 3. The mean age reported for the observation of nevus anemicus in this subgroup was 3.1 (3.6) years (range, 3 months–14 years). Fourteen of the patients developed nevus anemicus before skinfold freckling (Table 3).
Age and Order of Appearance of Diagnostic Criteria in Patients with NA or JXG Observed Before Definitive Diagnosis Of Neurofibromatosis 1.
| Patient | Sex | Age and order of appearance of diagnostic criteria and NA | Age at definitive diagnosis | ||||
|---|---|---|---|---|---|---|---|
| First | Second | Third | Fourth | Fifth | |||
| 1 | F | CALMs (18 mo) | Freckling (2 y) | NA (2 y) | Lisch nodules (7 y) | NF1 mutation (7 y) | 7 y |
| 2 | M | CALMs (5 mo) | NA (18 mo) | Freckling (2 y) | NF1 mutation (3 y) | – | 3 y |
| 3 | M | CALMs (6 mo) | NA (6 mo) | Plexiform NF (US) (1 y) | NF1 mutation (1 y) | – | 1 |
| 4 | M | CALMs (6 mo) | Freckling (6 mo) | NA (9 y) | NF×2 (9 y) | NF1 mutation (9 y) | 9 |
| 5 | F | CALMs (7 mo) | NA (7 mo) | NF1 mutation (1 y) | – | – | 1 |
| 6 | F | CALMs (9 mo) | NA (9 mo) | Lisch nodules (2 y) | NF1 mutation (3 y) | Freckling (3 y) | 2 |
| 7 | M | CALMs (6 mo) | Freckling (8 mo) | NA (9 mo) | NF1 mutation (1 y) | – | 1 |
| 8 | M | CALMs (4 y) | NA (4 y) | NF1 mutation (4 y) | Freckling (4 y) | – | 4 |
| 9 | M | CALMs (3 mo) | NA (3 mo) | NF1 mutation (1 y) | Freckling (7 y) | Plexiform NF (7) | 1 |
| 10 | F | CALMs (4 mo) | Freckling (1 y) | NA (3 y) | Plexiform NF (US) (4 y) | NF1 mutation (4 y) | 4 |
| 11 | M | CALMs (9 mo) | Freckling (NK) | NA (5 y) | NF1 mutation (7 y) | – | 7 |
| 12 | M | CALMs (6 mo) | NA/JXG (9/18 mo) | NF1 mutation (12 mo) | Freckling (18 mo) | Optic pathway glioma (2 y) | 1 |
| 13 | F | CALMs (2 y) | NA (9 y) | Freckling (10 y) | NF1 mutation (11 y) | – | 11 |
| 14 | F | CALMs (6 mo) | Freckling (NK) | NA (5 y) | NF1 mutation (7 y) | NF×2(US) (8 y) | 7 |
| 15 | F | CALMs (12 mo) | NA (12 mo) | NF1 mutation (2 y) | NF×2 (4 y) | – | 2 |
| 16 | M | CALMs (6 mo) | NA (6 mo) | NF1 mutation (1 y) | – | – | 1 |
| 17 | F | CALMs (9 mo) | NA/JXG (9/11 mo) | NF1 mutation (1 y) | – | – | 1 |
| 18 | F | CALMs (3 mo) | JXG/NA (18 mo) | NF1 mutation (6 y) | Lisch nodules (6 y) | NF×2 (6 y) | 6 |
| 19 | M | CALMs (1 y) | NA (1 y) | NF1 mutation (4 y) | Freckling (5 y) | – | 4 |
| 20 | M | CALMs (1 y) | Freckling (5 y) | NA (5 y) | NF×2 (US) (6 y) | – | 6 |
| 21 | M | MCCL (NK) | Freckling (NK) | NA (14 y) | NF×2 (US) (15 y) | – | 15 |
| 22 | F | CALMs (2 mo) | NA (3 mo) | Lisch nodules (12 y) | – | – | 12 |
Abbreviations: CALMs, café-au-lait macules; F, female; JXG, juvenile xanthogranuloma; M, male; NA, nevus anemicus; NF, neurofibroma; NK, not known; US, ultrasound.
CALMs are the hallmark diagnostic feature of NF1 as well as the main reason for suspecting the disease in the first months of life. These lesions can fade and even disappear completely in elderly patients,8 and they are not necessarily present in patients with familial spinal NF1.9 All the patients in our series, including those younger than 2 years, had at least 6 CALMs. According to estimates, between 66% and 99% of patients with NF1 will develop 6 or more CALMs in the first year of life.10,11 Observation by a primary care pediatrician of multiple CALMs in a newborn should prompt referral for evaluation by a dermatologist. In patients without a family history of NF1, this is when the journey to receiving a definitive diagnosis begins.11 The diagnostic delay has been estimated at 2 to 3 years,12 and just 20 to 46% of children without a family history of NF1 are diagnosed before 2 years of age.3,7 In our series, just 6.48% of patients met the diagnostic criteria for NF1 before this age. Some authors consider that observation of “typical” CALMs, with a round or oval morphology, a homogeneous brown color, well-defined regular borders, and a diameter larger than 5mm in the first months of life has a high positive predictive value for NF1.13 In the absence of other findings, most physicians opt for clinical follow-up or order a molecular study, which can be costly and take time. In our series, patients without a family history of NF1 were diagnosed on average 3 years and 8 months later than those with a history. These delays can be distressing for parents and also result in late detection of complications.3
The vast majority of patients with NF1 have freckles in childhood. Reported rates range from 85.3% to 93.7%,5,6 although a rate of just 21.1% was described in a retrospective analysis.7 In our series, 87.5% of our patients had freckling, although this feature only influenced diagnosis in 1 patient, who developed freckles before CALMs. Many authors believe that freckles should not be considered an independent diagnostic criterion for NF1, as they see them as being small CALMs with identical histopathologic features. The diagnosis of other genetic disorders involving CALMs and freckling, such as Legius syndrome, constitutional mismatch repair deficiency syndrome, and Noonan syndrome with multiple lentigines (formerly LEOPARD syndrome), is based solely on pigmentary criteria.14
In our series, the mean age at diagnosis (3.94 years) falls within the range reported in the literature (2.65–4.5 years),3,5,6 even though our inclusion criteria stipulated a third criterion or confirmation of an NF1 mutation in patients with CALMs and freckling only. Although the likelihood of false positives in patients with pigmentary features is very low,4 we understand that Crowe's sign provides supportive but not confirmatory evidence of a diagnosis of NF1.15
Most of the diagnostic criteria for NF1 are not confirmed until children are of school-going age.11 In the case of freckling, neurofibromas, and Lisch nodules, the likelihood of meeting the diagnostic criteria increases with time. Optic pathway gliomas and hyperintense signals on magnetic resonance imaging are more common in children aged 2–8 years. Paradoxically, findings that manifest early and have a high positive predictive value and are potentially of greater diagnostic utility are not considered to be diagnostic criteria by some experts.2 Examples are juvenile xanthogranuloma and nevus anemicus, which are more common in children under 2 years of age.14 Previous findings by our group have shown that all children who developed nevus anemicus (n=22) or juvenile xanthogranuloma (n=3) were subsequently diagnosed with NF1.14 In addition, the mean age at which nevus anemicus was detected was considerably lower than the mean age of diagnosis (3.1 vs. 3.94 years) and even lower than that of axillary freckling.
Neurofibromas are the third most common cutaneous finding in NF1. They are benign tumors that contain all the cellular components of the peripheral nerve. Retrospective studies of children with NF1 have shown that 38.1 to 38.4% have 2 or more cutaneous or subcutaneous neurofibromas and that 23 to 24.7% have 1 plexiform neurofibroma.5,7 These rates are slightly higher than those observed in our series: 37.19% and 20.37% respectively. An additional 20.37% of patients had a single lesion consistent with a neurofibroma. These lesions were not considered a diagnostic criterion, as they were isolated or missing confirmation of a plexiform pattern.
Because neurofibromas in children exhibit significant clinical variability, we decided to characterize and classify them using 9 clinical-ultrasound patterns detected using cluster analysis and associated with higher interobserver correlation.16 The easiest patterns to recognize are “classic” cutaneous neurofibromas, which are typically small, flesh-colored or slightly hyperpigmented papules or nodules with a soft consistency that acquire a sessile or pedunculated appearance as they develop.17 They are usually very evident after 6 years of age,11 although small papules or faint circumscribed elevations with normal pigmentation may be observed at earlier ages. These findings could help establish an earlier diagnosis via skin ultrasound or biopsy in doubtful cases.16
Although few studies have reported on the manifestations of NF1 during the perinatal period,18 early identification of plexiform neurofibromas would aid the diagnosis of NF1, even in the first months of life.19 Plexiform neurofibroma, a clinicopathologic entity in its own right, is a congenital lesion considered pathognomonic for NF1.20–22 Large CALMs with irregular borders that are present at birth or develop during the first months of life are suggestive of plexiform neurofibroma. Their detection facilitates early diagnosis via the use of biopsy or ultrasound to distinguish between the poorly named atypical CALMs and congenital melanocytic nevi.16,21 Like Peltonen et al.,23 we believe that neurofibroma types should be confirmed histologically or by ultrasound.
Dermatologists are in a position to detect other diagnostic criteria of NF1, such as Lisch nodules, which are an important diagnostic finding. In our series, observation of these nodules confirmed the diagnosis in 22% of patients. Iris hamartomas usually develop after 5 years of age, and their identification by slit lamp examination depends on the experience of the pediatric ophthalmologist and the collaboration of the patient. Test performed in younger children have lower sensitivity and specificity.5 Lisch nodules are also easily visualized by dermoscopy in patients with light-colored eyes.
The estimated frequency of optic pathway glioma lies between approximately 5% and 15%.24 Optic pathway gliomas were observed in 25.93% of the patients in our series, and they confirmed a diagnosis of NF1 in 14.81%. We believe that the high prevalence observed in our series is due to the pediatric neurology protocol in place at our unit, which contemplates the use of magnetic resonance imaging in asymptomatic patients with pigmentary features of NF1 after 2 years of age.
Osseous lesions are the least common finding in NF1. Although they can present at any age, the percentage of patients in our series did not differ from rates reported in the literature.25 Gait disturbances or curvature of the lower limbs should raise suspicion of bone alterations.
New molecular genetic diagnostic techniques such as next generation sequencing have a diagnostic accuracy of 95% in NF1,26 explaining why the inclusion of pathogenic variants as a diagnostic criterion has not generated controversy. In most cases, detection of specific mutations lacks prognostic value, although genotype–phenotype correlations can facilitate follow-up.27
Less than 25% of patients in our series had a family history of NF1, even though there is wide consensus that approximately 50% of all cases are hereditary.7 Parents familiar with the manifestations of NF1 probably do not visit a dermatologist or are not referred in the absence of comorbidities. Finally, we believe that the lower incidence of familial cases may be linked to genetic counseling and the use of preimplantation genetic diagnosis.28
The main limitations of this study are its retrospective, observational design and the small number of patients under 2 years of age. Another potential limitation is that our data on the age of presentation of different features of NF1 are based on clinical notes and may or may not coincide with the actual age of presentation or that reported by parents.
We agree with Legius et al.2 that it was necessary to update the NIH diagnostic criteria for NF1. In our opinion, however, the only change in the revised criteria that could help reduce diagnostic delays is the use of genetic tests to identify pathogenic NF1 variants.29 Genetic testing was already part of many routine diagnostic workups in NF,15,30 and based on our findings, its use in doubtful cases does not significantly reduce the mean age at diagnosis.7,14 We believe that the possibility of adding nevus anemicus and juvenile xanthogranuloma to the diagnostic criteria for NF1 should be revisited, as they are uncommon in other RASopathies, and in NF1 they usually appear within the first 2 years of life. We also believe that dermatologists should be invited to participate in multidisciplinary NF care teams.
FundingNo funding was received for this study. The thesis that contains part of the data reported in this study received the prize for second best thesis published in 2017 awarded by the regional group for Madrid, Castilla-La Mancha, and Extremadura (Sección Centro) of the Spanish Academy of Dermatology and Venereology (AEDV).
Conflicts of InterestThe authors declare that they have no conflicts of interest.
This study is part of the doctoral thesis entitled “Estudio clínico ecográfico de la neurofibromatosis tipo 1 en la edad pediátrica” (Clinical ultrasound study of neurofibromatosis type 1 in children).


