

INTRODUCCION
La fibromatosis hialina juvenil es una enfermedad extremadamente rara caracterizada por lesiones en la piel, hiperplasia gingival, contractura de las articulaciones y lesiones óseas1. Las lesiones de la piel consisten en múltiples tumores localizados con frecuencia en cuero cabelludo y alrededor de la nariz, pequeñas pápulas perladas y placas localizadas en tronco, mentón, orejas y alrededor de los orificios nasales. Se presenta un caso de fibromatosis hialina juvenil atendido en el Servicio de Dermatología del Hospital del Niño Jesús de Madrid.
DESCRIPCION DEL CASO
Una niña de 15 meses de edad, fruto de una primera gestación con concentraciones elevadas de alfafetoproteína en suero, de padres no consanguíneos, nació mediante cesárea a las 37 semanas de gestación, con peso al nacimiento de 2.720 g. La niña fue ingresada durante el período neonatal por presentar rigidez articular, ausencia de reflejos osteotendinosos y una importante limitación de los movimientos articulares. Tras los estudios complementarios realizados, incluyendo sistemático de sangre, bioquímica sérica, enzimas musculares, electromiograma, electroencefalograma, ecografía abdominal y cerebral y cribado auditivo y de metabolopatías, no se encontraron datos patológicos de interés salvo pequeños quistes en el área de la cisura tálamo-caudado carentes de significado patológico. La niña fue remitida a nuestra consulta para la valoración de una lesión tumoral de crecimiento progresivo en el labio superior que comenzó aproximadamente a la edad de 11 meses. La madre refería además unas lesiones de color rojizo presentes desde el nacimiento, localizadas en la cara posterior de cuello.
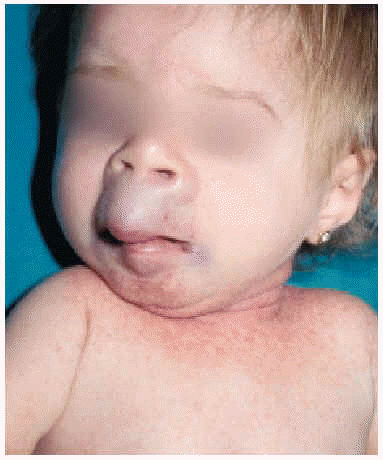
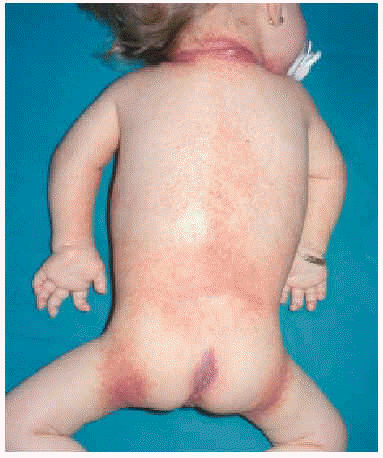
En la exploración física se apreció un grupo de pápulas perladas rosadas de pequeño tamaño alrededor de los orificios nasales y en cara posterior del cuello, con múltiples nódulos violáceos de consistencia firme, y de crecimiento lento y progresivo, en la región peribucal, en las orejas a la altura del hélix y alrededor del ano en zona sacrococcígea (figs. 1 y 2). Además se observaba una cierta asimetría facial, con deformidad de la nariz en silla de montar y una importante hipertrofia gingival que afectaba a la dentición. Las caderas, rodillas, tobillos, hombros, codos, muñecas y articulaciones interfalángicas de las manos estaban contracturadas en flexión con signos de tumefacción articular, y la columna vertebral carecía de las curvas fisiológicas. La paciente no ha logrado la deambulación, permaneciendo en actitud de flexión y aducción generalizada con rigidez vertebral. El examen neurológico fue difícil, pero no se observaron defectos funcionales.
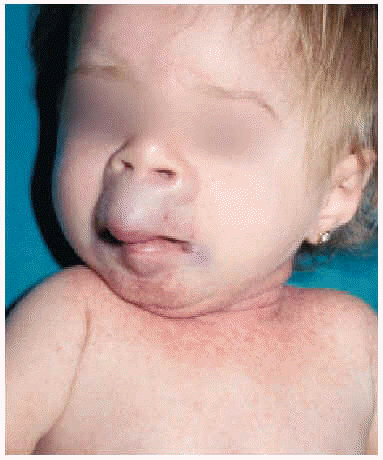
Fig. 1.--Nódulos violáceos en la región peribucal y en las orejas a la altura del hélix.
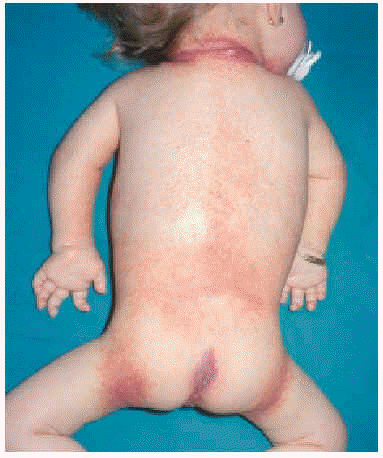
Fig. 2.--Nódulos violáceos alrededor del ano.
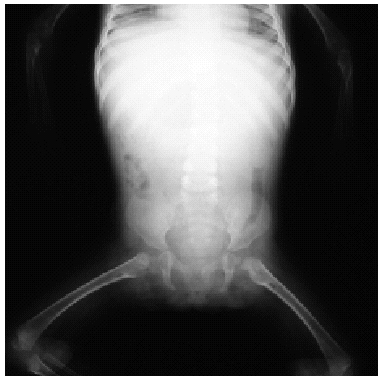
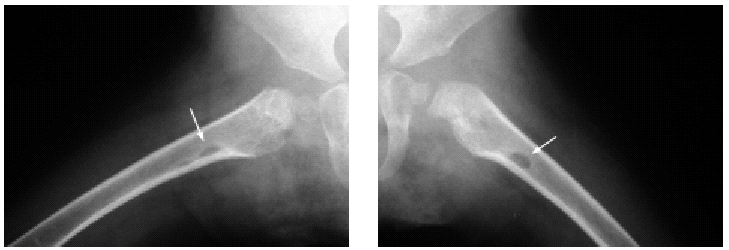
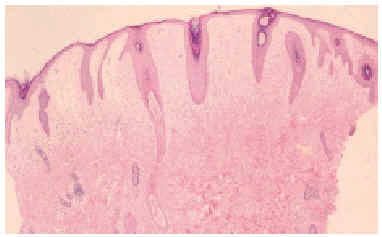
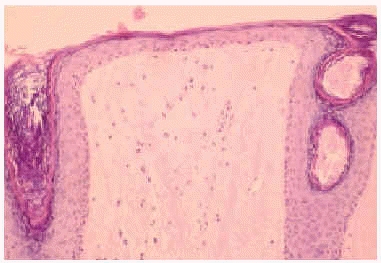
En el estudio complementario realizado que incluyó analítica completa, pruebas inmunológicas, niveles de aminoácidos y de mucopolisacáridos y pruebas serológicas para sífilis, citomegalovirus, toxoplasma y rubéola fueron normales. El cariotipo también fue normal y en el estudio radiológico se revelaron quistes osteolíticos en la parte proximal de ambos fémures y múltiples contracturas en flexión de las articulaciones con un aumento de la densidad de partes blandas periarticulares que les hacía radiológicamente visibles (figs. 3 y 4). El estudio histopatológico de una lesión tumoral puso de manifiesto una importante papilomatosis con una dermis papilar engrosada y una matriz eosinofílica hialina PAS+ sobre las que existen células de hábito fibroblástico (figs. 5 y 6). Existe una ausencia de tinción para fibras elásticas y un leve infiltrado inflamatorio linfocitario rodeando a algunos ductus.
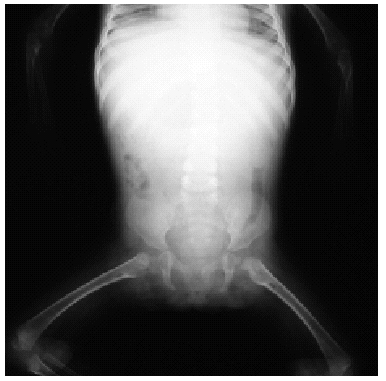
Fig. 3.--Aspecto radiológico de la paciente.
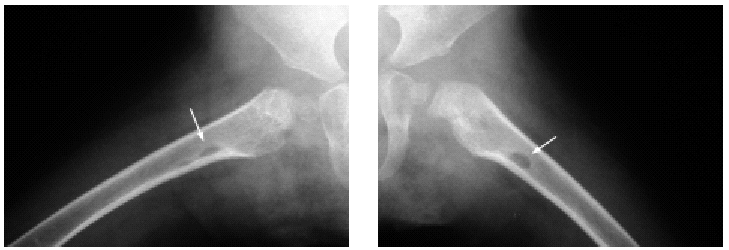
Fig. 4.--Estudio radiológico que muestra quistes osteolíticos en la parte proximal de ambos fémures.
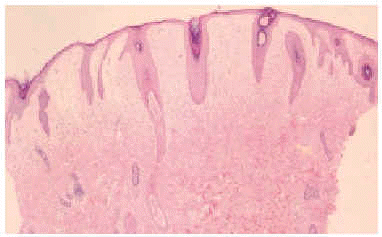
Fig. 5.--Estudio histopatológico de la lesión tumoral.
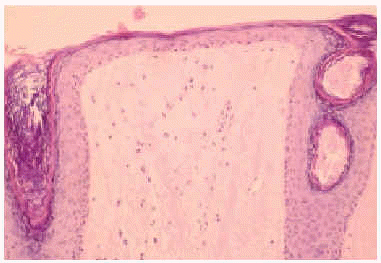
Fig. 6.--Estudio histopatológico de la lesión tumoral.
La paciente apenas mostró evolución pasados 6 meses de seguimiento, y persistieron las lesiones cutáneas y articulares. Su desarrollo neurológico fue adecuado, pero presentaba restricción casi absoluta de su motilidad articular. Falleció a los 2 años de edad por insuficiencia respiratoria desencadenada por una neumonía.
DISCUSION
La fibromatosis hialina juvenil se describió por primera vez en 1873 por Murray bajo el término molluscum fibroso2. Fue etiquetada posteriormente en 1967 como hialinosis sistémica3 hasta que más tarde en 1976, Kitano acuñó el término actual de «fibromatosis hialina juvenil»4. La fibromatosis hialina juvenil es una enfermedad hereditaria caracterizada por la producción y depósito de material hialino en el tejido conjuntivo de la piel y de las encías y menos frecuentemente en huesos y articulaciones5,6. Los múltiples casos descritos sugieren una herencia autosómica recesiva y también se han descrito casos de padres consanguíneos o historia familiar de fibromatosis hialina juvenil6-13. Recientemente se han descrito alteraciones homozigotas en el cromosoma 4q21 en estos pacientes14. La incidencia de esta enfermedad es baja; sólo se han descrito 65 casos y aparece más frecuentemente en niños pero también hay ejemplos publicados en adultos15.
La fibromatosis hialina juvenil se desarrolla precozmente; casi siempre lo hace en los primeros 4 años de vida, apareciendo el primer síntoma en la infancia precoz3,4,12 o incluso al nacimiento13,16. Las características clínicas son variables, pero las más frecuentes son las contracturas articulares y las lesiones en la piel y en el tejido subcutáneo. El primer síntoma y más constante suele ser la contractura articular, sobre todo de las rodillas y codos junto con caderas y tobillos. Raras veces falta la afectación articular12,17 o los cambios están limitados a una sola articulación18. Posteriormente aparecen las lesiones cutáneas, que de forma muy rara son el primer síntoma de la enfermedad16,18. Estas lesiones cutáneas son polimorfas y comprenden pequeñas pápulas blancoperladas, particularmente en cara y en nariz, pápulas grandes, alargadas y pequeños nódulos de apariencia translúcida con aspecto gelatinoso que se desarrollan en las orejas y en la nariz y, finalmente, nódulos firmes localizados en tronco, cuero cabelludo y en extremidades, que no ocurren en todos los pacientes. Estos tres tipos de lesiones cutáneas pueden permanecer en el mismo paciente3,9,16,18. La hiperplasia gingival es frecuente en la mayoría de los casos de fibromatosis hialina juvenil, de hecho, raras veces está ausente17,19 y produce con frecuencia una mala alineación dental que impide la correcta alimentación, como en nuestra paciente.
El retraso mental no es frecuente y son pacientes intelectualmente normales13. Las lesiones óseas radiológicas incluyen osteólisis en la parte distal de las falanges y el cráneo16, osteoporosis, erosiones simétricas en la cortical de huesos largos y calcificaciones de los tejidos blandos periarticulares y en la cápsula articular. La talla corta es una característica observada en algunos casos8. El resto de la exploración física es normal.
Gilaberte et al20 definieron los criterios clínicos aún vigentes para esta entidad (tabla 1). El grado de intensidad de la enfermedad es variable, en aquellos casos más graves presentan características superpuestas a la hialinosis sistémica infantil5,13,21; por ello, muchos autores sugieren que ambas enfermedades son el espectro de una misma entidad clínica8,22. La hialinosis sistémica infantil presenta una piel gruesa, dura y un compromiso visceral de curso progresivo que conlleva la muerte del paciente en los primeros años de vida.

Los aspectos histológicos de estos tumores de la piel son característicos y demuestran una abundante, homogénea y amorfa sustancia de apariencia condroide con células tumorales de tipo fibroblástico inmersas en esta sustancia eosinofílica PAS+. En las lesiones antiguas esta sustancia base es abundante y presumiblemente representa un precursor del colágeno. Al microscopio electrónico las células fusiformes son fibroblastos con un retículo endoplásmico rugoso dilatado y con vacuolas en su citoplasma, relleno de material granuloso o filamentoso23.
El tratamiento de la fibromatosis hialina juvenil es insatisfactorio. La escisión de las lesiones cutáneas, por motivos estéticos, conduce siempre a la recidiva local; además, son lesiones progresivas y raras veces regresan de forma espontánea17. Se obtiene una mejoría temporal de los síntomas articulares con corticoides sistémicos y hormona adrenocorticotropa. También se han utilizado el dimetilsulfóxido y el ketotifeno sin resultado24.
Durante el proceso de edición de este artículo se han identificado mutaciones en el gen de la proteína de morfogénesis capilar 2 (CMG2) en pacientes con fibromatosis hialina juvenil e hialinosis sistémica infantil (Hanks et al, Am J Hum Genet 2003; 73: 791-800).


