





INTRODUCCION
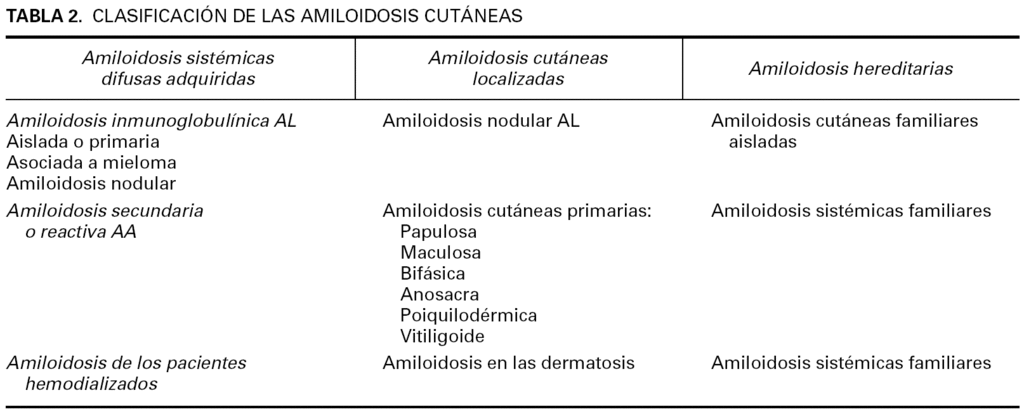
La amiloidosis es una enfermedad caracterizada por el depósito de amiloide en una o más localizaciones corporales, que puede llegar a afectar a todos los órganos. La sustancia amiloide es una proteína anómala, compuesta de fibrillas huecas, rígidas, lineales, no ramificadas, de 7,5 a 10 nm de diámetro, dispuestas en una red laxa con plegamiento de tipo β 1. La sustancia amiloide tiene dos componentes: el común y el específico. El componente común, presente en todas las sustancias amiloides de todas las amiloidosis, es una proteína denominada componente amiloide P (AP), que deriva del componente amiloide sérico (SAP), una proteína que de forma habitual se encuentra en el suero de los pacientes sanos. El componente común representa el 15-20 % del peso seco de la sustancia amiloide. El componente específico es una proteína que define el tipo de amiloidosis 2 (tabla 1). En todas las formas cutáneas, excepto en la amiloidosis cutánea nodular primaria, la sustancia amiloide deriva de los queratinocitos (tabla 2).

La amiloidosis cutánea nodular primaria (ACNP) es la forma menos frecuente de amiloidosis. Es una forma inmunoglobulínica, cutánea y primaria, es decir, en la piel existe un clon de células plasmáticas que fabrican cadenas ligeras de inmunoglobulinas que degeneran a sustancia amiloide de tipo AL. Es por lo tanto, un plasmocitoma extramedular 3. La edad de presentación suele ser hacia los 65 años. La ACNP es más frecuente en los hombres. Clínicamente se caracteriza por la aparición de nódulos, que pueden formar masas tumorales, de coloración ámbar y de consistencia blanda o compacta. Las lesiones se localizan con frecuencia en las piernas, aunque también pueden aparecer en la cara y el tronco 4-6. Esta forma clínica de amiloidosis puede evolucionar hacia una enfermedad sistémica, como un mieloma (15-50 % de los casos, según las series), y progresar a una amiloidosis sistémica 4-6. Entre las manifestaciones sistémicas que pueden presentarse figuran la afectación cardiaca (26-30 %), renal (11-32 %), sistema nervioso periférico (5-17 %), intestinal, articular (16 %), hepática (50 %) y esplénica, tal y como sucede en los casos de amiloidosis sistémica.
En cuanto a la histopatología, lo más frecuente es encontrar el depósito de sustancia amiloide, de tipo AL, en la dermis papilar, alrededor de los anejos, de la pared de los vasos, de los músculos erectores, de los folículos pilosos y de los adipocitos, formando los llamados «anillos amiloides».
Se describe el caso de una paciente con síndrome de Sjögren primario que desarrolló una ACNP en el abdomen.
DESCRIPCION DEL CASO
Una mujer de 69 años de edad con antecedentes personales de hipertensión arterial (HTA), vértigo posicional paroxístico, alergia a betalactámicos, colecistectomía, ovariectomía derecha y hernia discal L4-L5, estaba en tratamiento con omeprazol, losartán, tramadol y topiramato.
En el año 1997, la paciente acudió a la consulta de medicina interna al padecer episodios periódicos de dolor e inflamación en la glándula parótida derecha, sequedad ocular, oral y genital, astenia, pérdida de 10 kg de peso, dolores osteomusculares generalizados y parestesias generalizadas nocturnas. Con la sospecha clínica de síndrome seco, se le practicó una biopsia de glándulas salivales menores (labio inferior). La histopatología puso de manifiesto una sialoadenitis crónica inespecífica, sin signos de malignidad, con una tinción de tioflavina para amiloide negativa. El estudio se completó con una analítica que mostró anticuerpos antinucleares (ANA) positivos a título de 1/640, con anti-SSA/Ro de 47 U/ml (VN: 0-25), anti-SSB/La de 41 U/ml (VN: 0-25). La paciente fue entonces diagnosticada de síndrome de Sjögren primario.
Acudió a nuestra consulta por presentar, desde hacía 10 años, por lo tanto 3 años antes de ser diagnosticada de síndrome de Sjögren primario, una lesión de crecimiento lento, pruriginosa, en la región epigástrica. Había recibido tratamientos con corticoides tópicos de potencia alta sin mejoría, y en aquel momento no se realizó estudio histopatológico. A la exploración física se apreciaba una placa de aspecto nodular, eritematoparduzca, de bordes bien delimitados, infiltrada y no dolorosa a la palpación, de 10 cm de diámetro, localizada en la región epigástrica (fig. 1).
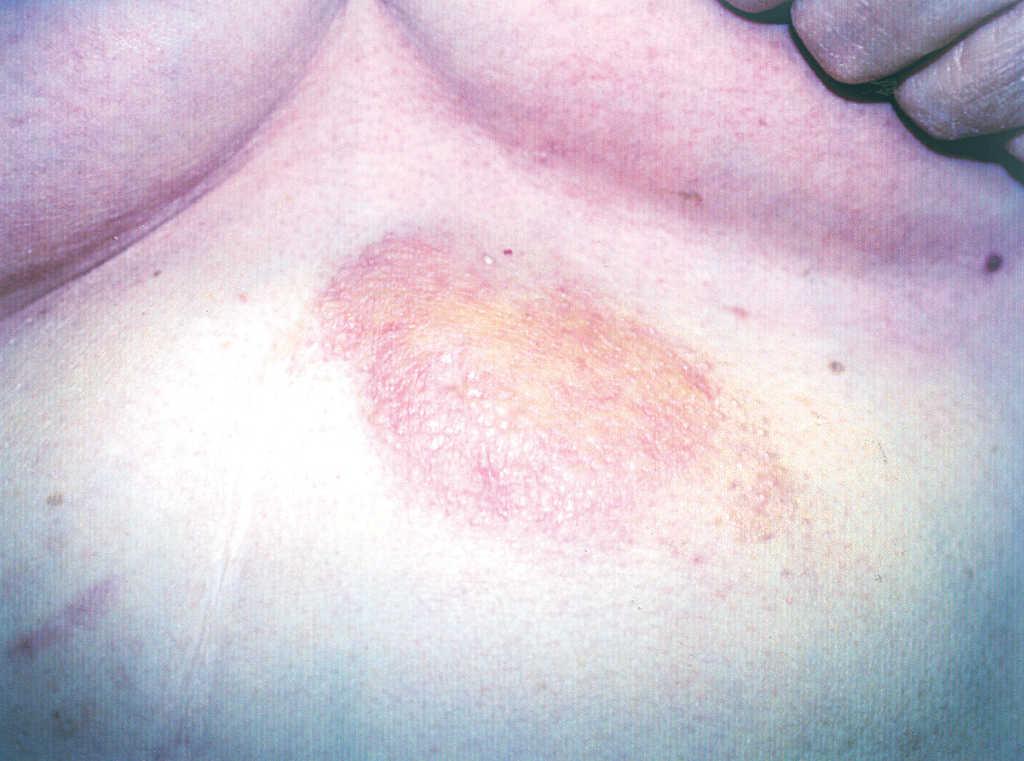
Fig. 1.--Placa nodular amarillenta en región epigástrica.
El hemograma, la velocidad de sedimentación globular (VSG), proteína C reactiva (PCR), coagulación, complemento, serología de virus de la hepatitis B y C (VHB y VHC), vitamina B12, ácido fólico, hormonas tiroideas, biomarcadores tumorales, inmunoglobulinas y orina (sedimento e inmunoelectroforesis) no mostraron alteraciones significativas. Las proteínas totales estaban aumentadas: 8,8 g/dl (VN: 6,6-8-3). El estudio electroforético de las proteínas evidenció un aumento de la gammaglobulina: 31,6 % (VN: 12-19 %). No obstante, los estudios de paraproteinemia, tanto en suero como en orina, fueron negativos. El factor reumatoide estaba elevado a 414 U/ml (0-20), y el estudio de autoinmunidad mostró ANA a títulos de 1/640, con patrón granular, anti-SSA/Ro superior a 100 U/ml (VN: 0-25), anti-SSB/La de 99,63 U/ml (VN: 0-25), con el resto de autoanticuerpos dentro de los límites de la normalidad. El estudio histopatológico de la lesión puso de manifiesto una acumulación de material eosinófilo, amorfo y acelular, en los dos tercios superiores de la dermis. Este material se encontraba también depositado en la dermis más profunda a nivel perivascular y perianexial, en donde también existía un discreto infiltrado de células plasmáticas (fig. 2). Las tinciones con rojo Congo y tioflavina fueron positivas (fig. 3), pero el material no se teñía con la proteína A del amiloide. Sin embargo, las tinciones con antisueros de cadenas ligeras κ y λ fueron positivas, demostrando la existencia de células plasmáticas policlonales.
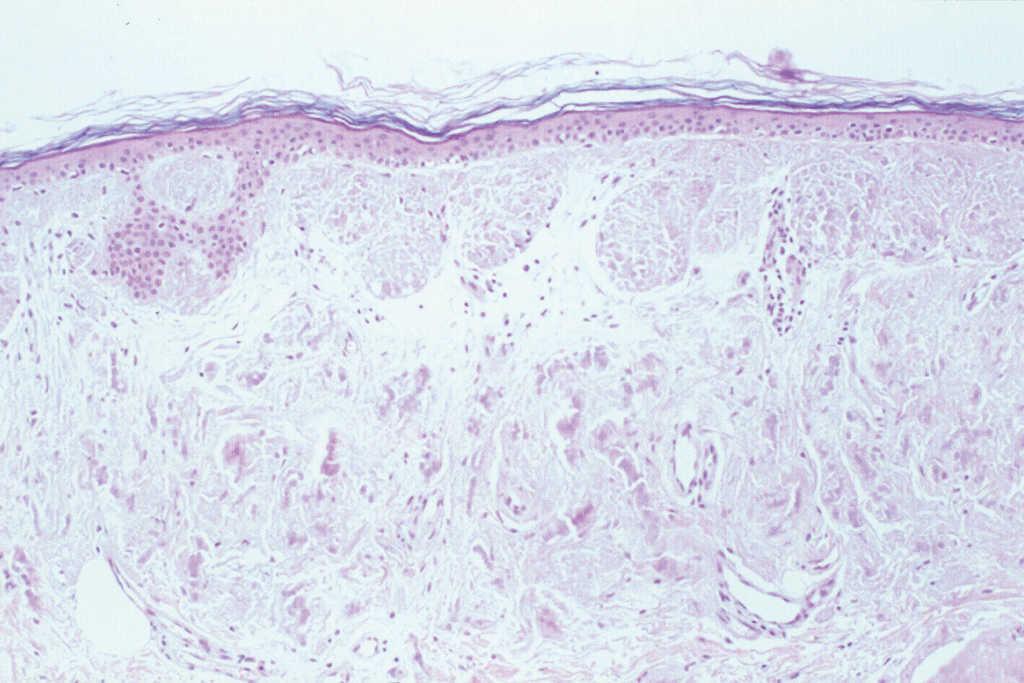
Fig. 2.--Presencia de acumulaciones de un material eosinófilo, acelular y amorfo en la dermis, con presencia de células plasmáticas. (Hematoxilina-eosina, x20.)
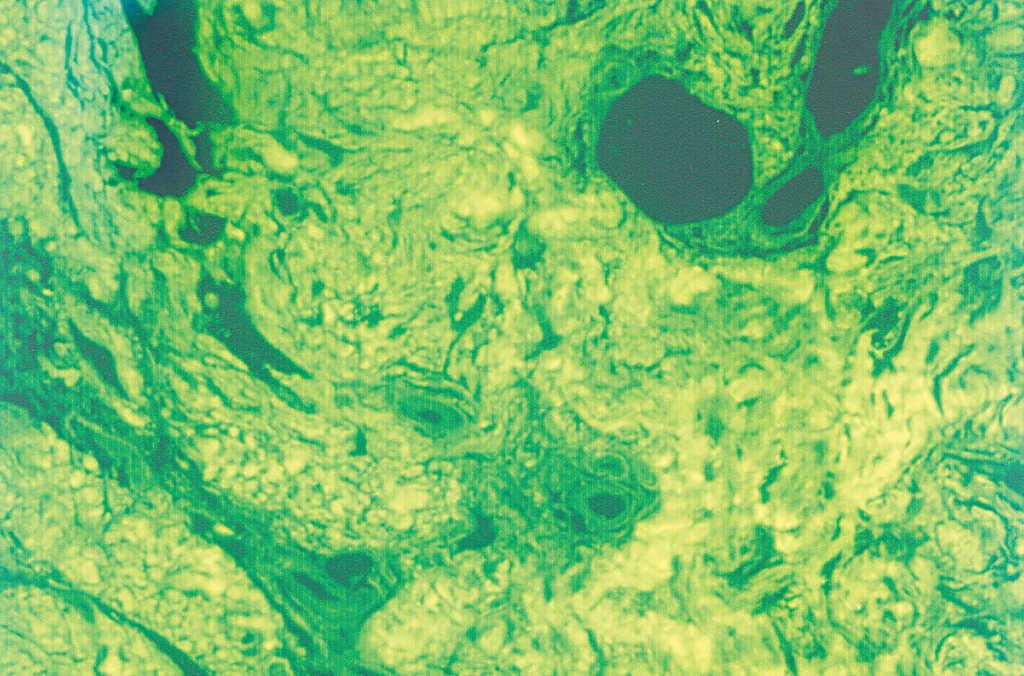
Fig. 3.--Con el microscopio de fluorescencia se aprecia el depósito de sustancia amiloide. (Tinción de tioflavina, x 40.)
DISCUSION
Desde el año 1971 se han publicado menos de 20 casos de ACNP, y la mitad se han manifestado en pacientes con síndrome de Sjögren primario 7,8. Entre las causas del llamado síndrome seco se debe descartar la infiltración amiloide de las glándulas salivales y lagrimales, especialmente si los autoanticuerpos anti-SSA y anti-SSB son negativos 7. No obstante, pacientes con serologías positivas han desarrollado previamente, simultánea o posteriormente, manifestaciones cutáneas compatibles clínica e histopatológicamente con ACNP. Estos casos no son amiloidosis secundarias al síndrome de Sjögren, ya que en todos ellos el depósito de amiloide reaccionaba con los anticuerpos contra AL (cadenas ligeras de inmunoglobulinas λ y/o κ) 8-11.
Algunos estudios inmunohistoquímicos demuestran que el citoplasma de las células plasmáticas se tiñe con AL, tanto de tipo κ como λ, por lo que las cadenas ligeras de inmunoglobulinas policlonales serían el origen del depósito amiloide en los casos de ACNP, tratándose entonces de una enfermedad reactiva más que neoplásica. No obstante, hay autores que han demostrado, tanto por inmunohistoquímica como por PCR, la monoclonalidad de las células plasmáticas, así como de la sustancia amiloide 3,12.
El tratamiento de las lesiones cutáneas de la ACNP engloba la cirugía, la dermoabrasión con láser de colorante pulsado y láser de CO2. Se han utilizado los corticoides potentes en cura oclusiva, los antihistamínicos en caso de intenso prurito, la asociación de corticoides tópicos con dimetil sulfóxido (DMSO), ácido retinoico al 0,05 %, ácido tricloroacético al 33 %, fototerapia con UVB y calcipotriol tópico, así como la crioterapia, el curetaje y la cauterización. Sin embargo, los resultados obtenidos con todos estos tratamientos son muy variables.
La ACNP puede presentar un curso benigno durante años; sin embargo, se debe tener en cuenta que algunos pacientes desarrollan más tarde una paraproteinemia con amiloidosis sistémica evidente.


