


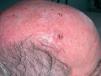


HISTORIA CLINICA
Varón de 83 años, con antecedentes personales de carcinoma epidermoide de laringe intervenido en 1998 con vaciamiento ganglionar cervical completo y radioterapia coadyuvante, diabetes mellitus no dependiente de insulina, úlcera duodenal y psoriasis, que acude a la consulta de dermatología por presentar, desde hace 2 meses, un tumor perlado en hemifrente derecha, ulcerado en su parte central que ha aumentado progresivamente hasta alcanzar el tamaño actual. No le causa ningún tipo de sintomatología subjetiva.
EXPLORACION FISICA
Tumor globuloso, perlado, con costra serosa en la parte central de 2,5 cm de diámetro situado en hemifrente derecha (fig. 1). No se palpaban adenopatías locorregionales. El resto de la exploración por órganos y aparatos no mostró alteraciones significativas.
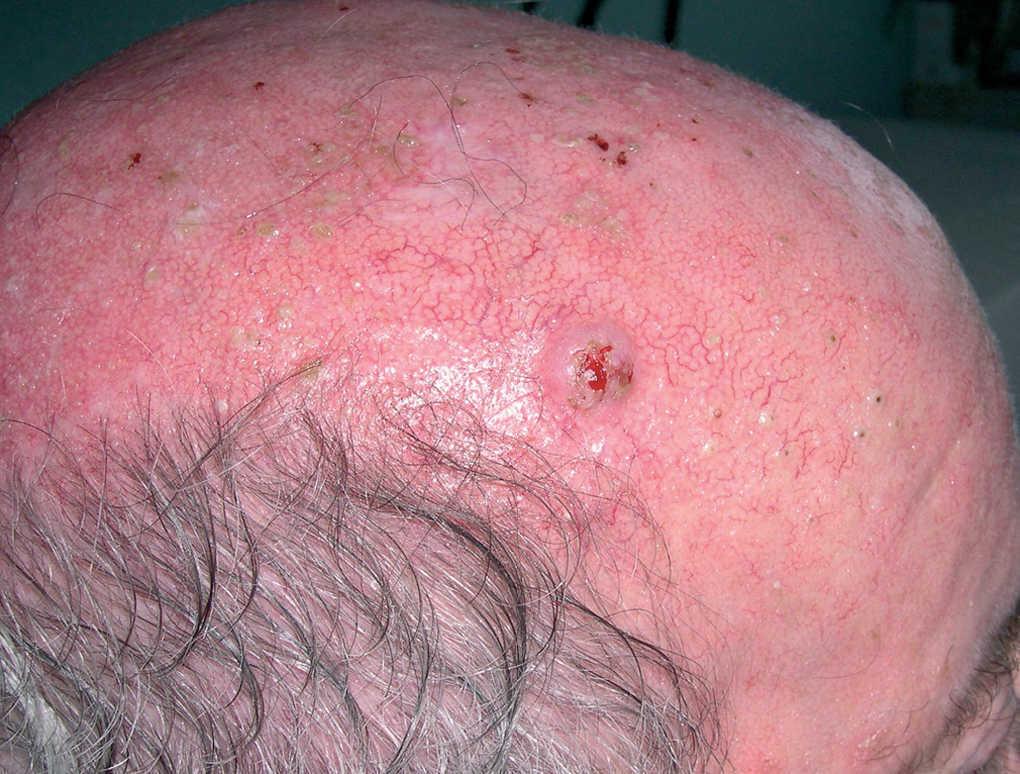
Fig. 1.--Nódulo perlado en hemifrente derecha de rápido crecimiento.
EXPLORACIONES COMPLEMENTARIAS
Los exámenes complementarios solicitados: hemograma, bioquímica general, sedimento urinario, tomografía computarizada cervicofacial y toracoabdominal y marcadores tumorales ofrecieron resultados dentro de la normalidad. Se realizó extirpación biopsia de la lesión con márgenes quirúrgicos de 0,5 cm en todas direcciones (figs. 2 y 3).
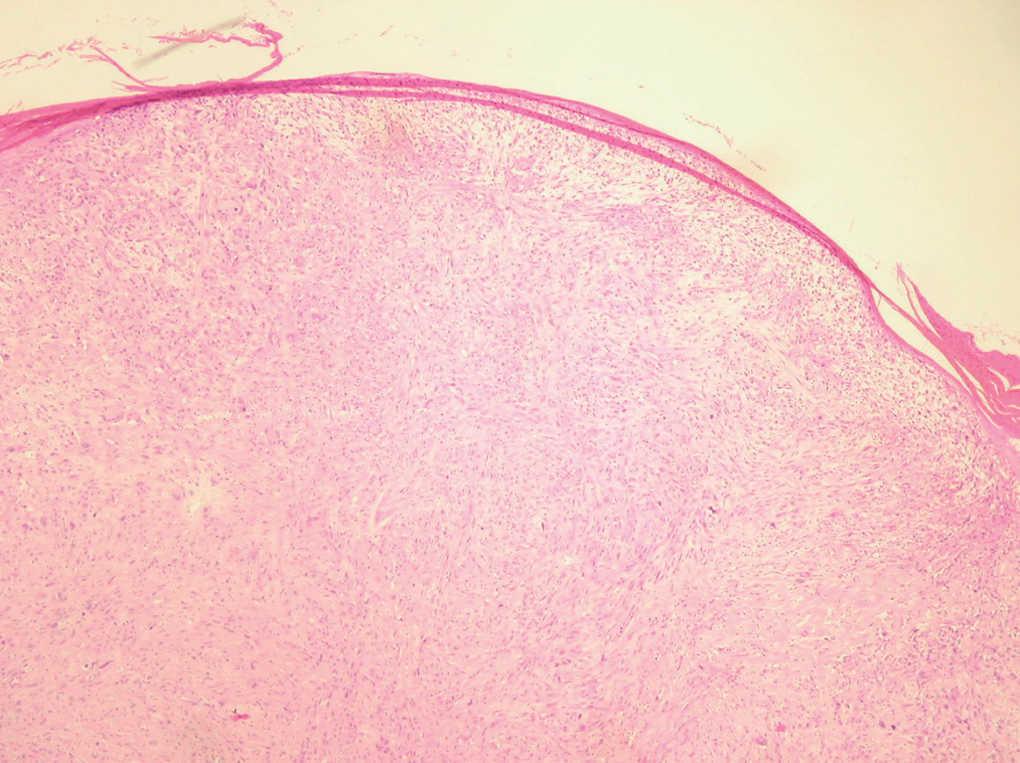
Fig. 2.--Neoformación de estirpe mesenquimatosa constituida por células fusiformes. (Hematoxilina-eosina, x10.)
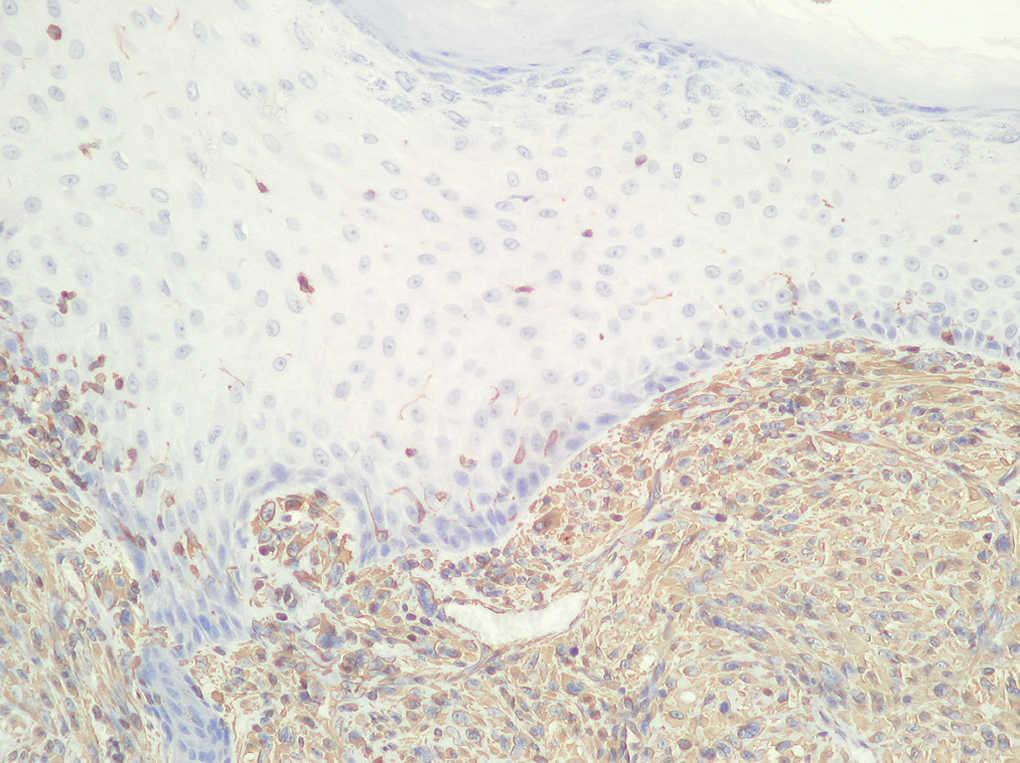
Fig. 3.--Estudio inmunohistoquímico. (Vimentina positiva.)
DIAGNOSTICO
Histiocitoma fibroso maligno (HFM) (variedad pleomórfica-estoriforme).
HISTOPATOLOGIA
Neoformación de estirpe mesenquimatosa constituida por células de citoplasma y núcleo fusiforme, dispuestas en áreas y fascículos, entre las que aparecen células multinucleadas de cuerpo extraño (fig. 2), con focos de necrosis y elevada actividad mitótica. Las técnicas inmunohistoquímicas realizadas mostraron negatividad para actina, CD34, HMB45, EMA, CD68, CAM 5.2., desmina, pancitoqueratinas, S-100 y positividad para vimentina (fig. 3).
EVOLUCION Y TRATAMIENTO
Con el diagnóstico histológico se procedió a realizar ampliación de márgenes quirúrgicos y extirpación de la lesión con 2 cm de margen de seguridad sin que se hayan producido recidivas locales ni metástasis a distancia a los 9 meses de la intervención inicial.
COMENTARIO
Denominado por primera vez en 1964 como xantoma fibroso maligno y posteriormente en 1972 como HFM, es el sarcoma más común de los tejidos blandos en la edad adulta tardía, y es poco frecuente la forma de presentación cutánea. Suele afectar a los tejidos blandos profundos y al músculo estriado de las partes proximales de las extremidades, y el retroperitoneo es una de sus localizaciones más frecuentes.
Clínicamente se diferencian dos variantes de HFM: superficial, que envuelve el tejido subcutáneo por encima de la fascia y que raramente afecta la piel, y profundo, que abarca los tejidos por debajo de la fascia y puede también envolver el tejido muscular 1,2. Normalmente se presenta como un nódulo blanco-grisáceo, de tamaño variable donde suele ser frecuente encontrar áreas focales de hemorragia y necrosis. La mayoría se encuentran en las extremidades, y un tercio de ellos se localizan en los muslos y las nalgas. Es más frecuente en varones que mujeres y predomina en la franja de edad entre los 50 y 80 años, aunque la variante histológica angiomatoide se observa en pacientes jóvenes de entre 15 y 25 años de edad con relativamente buen pronóstico.
Se han considerado como factores predisponentes 3 las zonas de radiodermitis 2, úlceras crónicas, cicatrices de quemaduras o incluso aparición del tumor sobre cicatrices de vacunación. También se ha relacionado con lesiones óseas benignas (infartos óseos, enfermedad de Paget), osteomielitis crónica 4 y lupus eritematoso discoide.
La histogénesis de este tumor maligno es incierta y controvertida. La tendencia actual es creer que la célula progenitora no es un fagocito mononuclear sino una célula mesenquimatosa poco definida que puede diferenciarse en las líneas histiocíticas y fibroblástica 5. Recientes estudios 6,7 han mostrado la presencia de un tipo de célula específica, la célula fibrohistiocítica, que está presente en varios procesos crónicos inflamatorios. El papel de esta célula en la génesis del tumor podría estar justificado por la observación frecuente de una asociación entre el HFM y los procesos inflamatorios crónicos citados.
Se han reconocido 5 subtipos desde el punto de vista histológico: pleomórfico-estoriforme, mixoide, de células gigantes, inflamatorio y angiomatoide. Entre estos subtipos existen características superpuestas y se pueden encontrar dos áreas diferentes del tumor con dos subtipos diferentes.
La variante pleomórfico-estoriforme correspondiente a nuestro caso es la forma más común. Está formada por una mezcla de células fusiformes redondeadas, grupos o láminas de histiocitos y células gigantes multinucleadas dispersas. Son bastante comunes los cambios mixoides focales y la presencia de mitosis. A veces se forma material metaplásico condroide u osteoide, células de xantoma y siderófagos.
En el estudio inmunohistoquímico las células tumorales son vimentina positivas y presentan fuerte tinción para CD74 (LN-2). Puede demostrarse varios marcadores de histiocitos mediante el uso de la inmunoperoxidasa en un 60-80 % de los HFM. Los marcadores usados incluyen α1-antitripsina y α1-antiquimiotripsina. El CD68 (KP-1) es con frecuencia positivo, mientras que el CD34 suele ser negativo.
El HFM es raramente diagnosticado previa extirpación y estudio patológico y se ha de realizar diagnóstico diferencial con fibroxantoma atípico, tumor dérmico más superficial y de menor tamaño comparado con el HFM, posiblemente una variante superficial o incluso una forma inicial del HFM 7; sarcoma epitelioide, que comparte con el HFM la presencia de células espinosas y poligonales, carece de células grandes, multinucleadas y abigarradas; dermatofibroma subcutáneo y profundo, y dermatofibrosarcoma protuberans en el que a diferencia del HFM la tinción para CD34 normalmente es positiva.
La importancia de este tumor radica en que puede ser curado si se trata de forma precoz y correctamente. El tratamiento de elección consiste en realizar una amplia extirpación quirúrgica y se observan menos recidivas mediante cirugía de Mohs. La radioterapia y la quimioterapia pueden utilizarse como coadyuvantes de la cirugía o como medida paliativa y pueden mejorar el pronóstico.
El pronóstico en general es malo, con una supervivencia media a los 5 años del 15-30 %. Parece depender del grado histológico, el tamaño y la localización del tumor 8. En relación con el tamaño, el pronóstico es mejor cuanto menor es su diámetro y cuanto más superficialmente está localizado.
La variante angiomatoide es la de mejor pronóstico y la inflamatoria la que mayor probabilidad tiene de generar metástasis a distancia. Son comunes las recidivas locales y las metástasis. Finalmente, en relación con la localización 9, los tumores confinados en tejido subcutáneo que no afectan la fascia raramente metastatizan (< 10 % de casos); si afectan la fascia pero no infiltran el tejido muscular, metastatizan en un 27 % de casos; y si culmina por afectar el plano muscular aumenta el porcentaje hasta el 43 %.
Pezzi et al 10,11 aportan que la mayoría de metástasis del HFM ocurren en pulmón (90 %), seguido de ganglios linfáticos (12 %), hueso (8 %) e hígado (1 %). Tienen especialmente mal pronóstico los que asientan en zonas de radiodermitis.
Weiss y Enzinger 12 observan que después de la extirpación sólo un 32 % de casos presentan curación, un 42 % desarrolla metástasis y un 44 % de casos presentan recurrencia local. La mayoría de muertes ocurre dentro de los 2 años posteriores al diagnóstico inicial.


