



Keratoacanthomas (KAs) are usually sporadic and solitary lesions appearing in patients on sun-exposed areas. Chronic sun-exposure, imiquimod, BRAF inhibitors and hedgehog pathway inhibitors can be triggering agents. Genetic causes of KAs such as Ferguson-Smith syndrome (multiple self-healing squamous epitheliomas -MSSE) and generalized eruptive keratoacanthoma (Grzybowski syndrome) are characterized by the development of dozens to hundreds of KAs at a young age and are not associated with other cutaneous or extracutaneous tumours1. Other associated genetic diseases are xeroderma pigmentosum (XP) and Muir-Torre syndrome (MTS).
We sought to describe the clinical-epidemiological characteristics and associated genetic disorders of young patients with KAs in our centers, and to develop a diagnostic algorithm. We describe three patients who developed KAs before age 40 and who harbored unrecognized genetic diseases predisposing to these cutaneous lesions.
Patient 1 was a 54-year-old male with no relevant family history and a personal history of intense sunlight exposure who had had more than ten KAs since age 39. He developed growing centrifugal lesions on the left forearm after radiotherapy for a solitary KA. The lesions were clinically and histologically suggestive of keratoacanthoma marginatum centrifugum (KMC) (Fig. 1). No loss of MMR immunohistochemical markers was observed in the tumors, and the lesions were successfully treated with intralesional 5-fluorouracil and oral retinoids. A screen analysis of the TGFBR1 gene and a likely pathogenic c.301 T>C heterozygous variant was detected.
.")
Patient 2 was an otherwise healthy 38-year-old male who developed a rapidly growing KA measuring 13 mm on the nose. His father had died at age 53 after developing metastatic colon cancer. MMR immunohistochemical showed loss of MSH2 and MSH6. The lesion was treated with intralesional methotrexate, showing complete regression in a 12-week period. A constitutional c.142G>T heterozygous mutation in the MSH2 gene (predicted to generate a truncating protein, p.Glu48*) was identified by means of a next generation sequencing panel and a Sanger sequencing validation, confirming the diagnosis of Lynch syndrome.
Patient 3 was an otherwise healthy male with no family history who developed a 17 mm diameter KA in the cervical region at age 38. The lesion was surgically excised and MMR immunohistochemical stains showed loss of MSH2 and MSH6. A germinal MMR gene study showed a large deletion (c.(?_68)_1276+?del) involving exons 1 to 7 in the MSH2 gene, confirming the diagnosis of Lynch syndrome, MTS subtype. The patient developed a high grade pT2 carcinoma in the ureter at age 41. Several facial sebaceous adenomas and a KA in the cervical region have also been removed to date.
The development of solitary or multiple KAs at a young age may be the presenting sign of genetic disorders which may be associated with malignant neoplasms. Recognizing such associations is important in order to prevent possible complications.
MSSE, an autosomal dominant disease, is caused by mutations in the TGFBR1 gene together with permissive variants at a locus near the TGFBR1 locus on chromosome 92. Patients with MSSE develop multiple lesions which grow slowly and resolve within months, while new lesions appear. There is variable penetrance, ranging from asymptomatic carriers to patients developing lesions from the first to the seventh decade of life. Some patients may show KCM lesions, characterized by the development of coral reef-like lesions with progressive peripheral expansion and atrophic central healing. The development of KCM after radiotherapy, should suggest the diagnosis of MSSE3. Alternative treatment modalities such as oral retinoids, corticosteroids or intralesional/systemic methotrexate can be used.4
XP is associated with the early development of skin cancer but KAs are seldom the first tumor manifestation. MTS is reported in 10-30% of families with Lynch syndrome mainly harboring mutations of MSH2 or MLH1 genes5,6. MMR immunohistochemical markers should be assayed on tumor samples from patients with sebaceous cutaneous neoplasms, multiple KAs and/or appearing at a young age. However, the lack of expression of MMR proteins in sebaceous neoplasms is not exclusive to patients with MTS and some cases which maintain the expression of the MMR proteins have an underlying germline DNA repair defect.
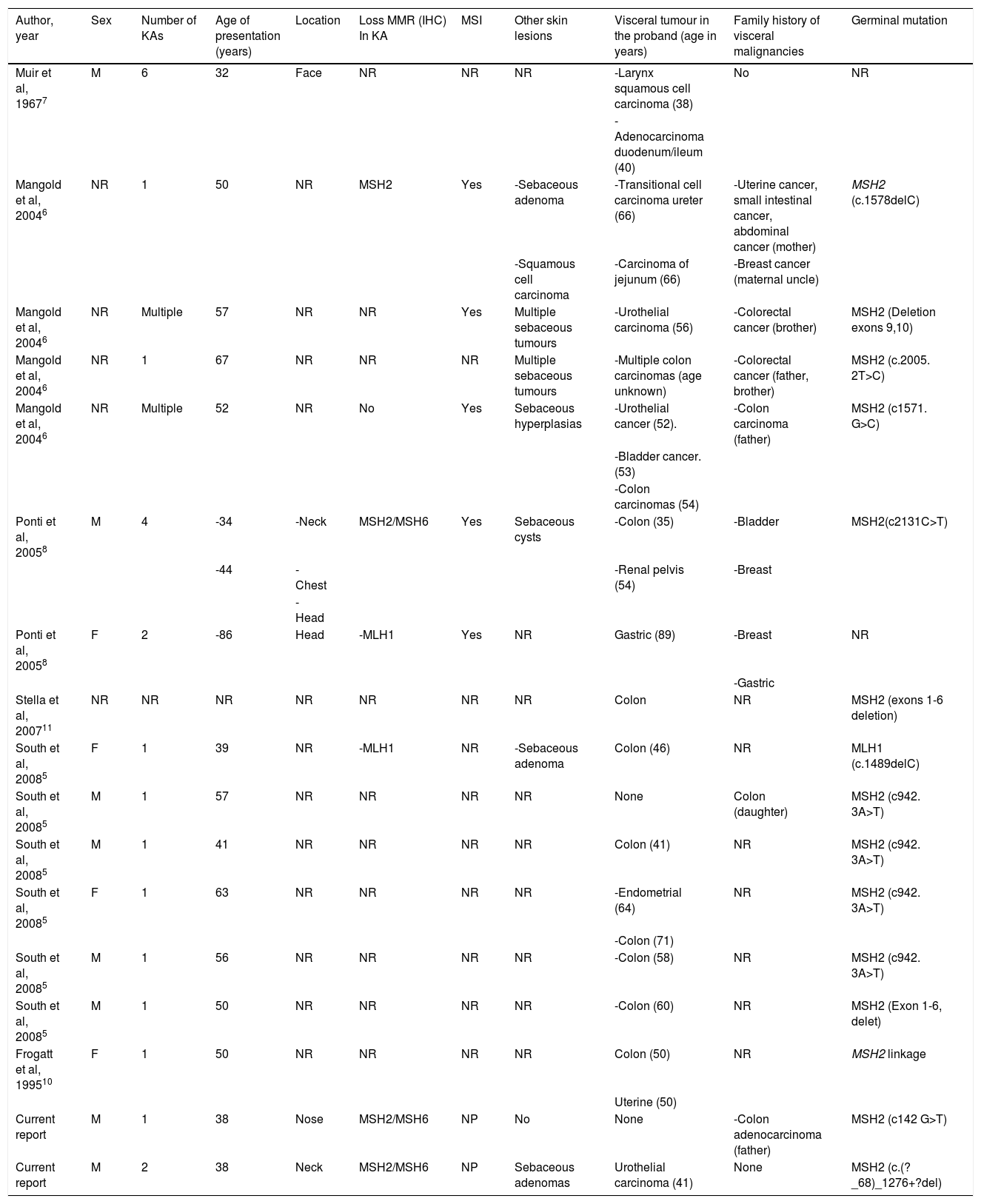
A literature review shows that approximately 89% of the patients with MTS-associated KAs harbor MSH2 mutations, (Table 1), which is in accordance with the reported mutational prevalence of non-KA cutaneous neoplasms in MTS5–10. MTS-associated KAs7 are often indistinguishable histologically from sporadic KAs. KAs in patients with MTS usually develop before the diagnosis of visceral neoplasms is performed, while sebaceous tumors in MTS are commonly detected after the diagnosis of visceral malignancies.
Cases reported in the literature of patients with Muir-Torre syndrome-associated keratoacanthomas.
| Author, year | Sex | Number of KAs | Age of presentation (years) | Location | Loss MMR (IHC) In KA | MSI | Other skin lesions | Visceral tumour in the proband (age in years) | Family history of visceral malignancies | Germinal mutation |
|---|---|---|---|---|---|---|---|---|---|---|
| Muir et al, 19677 | M | 6 | 32 | Face | NR | NR | NR | -Larynx squamous cell carcinoma (38) | No | NR |
| -Adenocarcinoma duodenum/ileum (40) | ||||||||||
| Mangold et al, 20046 | NR | 1 | 50 | NR | MSH2 | Yes | -Sebaceous adenoma | -Transitional cell carcinoma ureter (66) | -Uterine cancer, small intestinal cancer, abdominal cancer (mother) | MSH2 (c.1578delC) |
| -Squamous cell carcinoma | -Carcinoma of jejunum (66) | -Breast cancer (maternal uncle) | ||||||||
| Mangold et al, 20046 | NR | Multiple | 57 | NR | NR | Yes | Multiple sebaceous tumours | -Urothelial carcinoma (56) | -Colorectal cancer (brother) | MSH2 (Deletion exons 9,10) |
| Mangold et al, 20046 | NR | 1 | 67 | NR | NR | NR | Multiple sebaceous tumours | -Multiple colon carcinomas (age unknown) | -Colorectal cancer (father, brother) | MSH2 (c.2005. 2T>C) |
| Mangold et al, 20046 | NR | Multiple | 52 | NR | No | Yes | Sebaceous hyperplasias | -Urothelial cancer (52). | -Colon carcinoma (father) | MSH2 (c1571. G>C) |
| -Bladder cancer. (53) | ||||||||||
| -Colon carcinomas (54) | ||||||||||
| Ponti et al, 20058 | M | 4 | -34 | -Neck | MSH2/MSH6 | Yes | Sebaceous cysts | -Colon (35) | -Bladder | MSH2(c2131C>T) |
| -44 | -Chest | -Renal pelvis (54) | -Breast | |||||||
| -Head | ||||||||||
| Ponti et al, 20058 | F | 2 | -86 | Head | -MLH1 | Yes | NR | Gastric (89) | -Breast | NR |
| -Gastric | ||||||||||
| Stella et al, 200711 | NR | NR | NR | NR | NR | NR | NR | Colon | NR | MSH2 (exons 1-6 deletion) |
| South et al, 20085 | F | 1 | 39 | NR | -MLH1 | NR | -Sebaceous adenoma | Colon (46) | NR | MLH1 (c.1489delC) |
| South et al, 20085 | M | 1 | 57 | NR | NR | NR | NR | None | Colon (daughter) | MSH2 (c942. 3A>T) |
| South et al, 20085 | M | 1 | 41 | NR | NR | NR | NR | Colon (41) | NR | MSH2 (c942. 3A>T) |
| South et al, 20085 | F | 1 | 63 | NR | NR | NR | NR | -Endometrial (64) | NR | MSH2 (c942. 3A>T) |
| -Colon (71) | ||||||||||
| South et al, 20085 | M | 1 | 56 | NR | NR | NR | NR | -Colon (58) | NR | MSH2 (c942. 3A>T) |
| South et al, 20085 | M | 1 | 50 | NR | NR | NR | NR | -Colon (60) | NR | MSH2 (Exon 1-6, delet) |
| Frogatt et al, 199510 | F | 1 | 50 | NR | NR | NR | NR | Colon (50) | NR | MSH2 linkage |
| Uterine (50) | ||||||||||
| Current report | M | 1 | 38 | Nose | MSH2/MSH6 | NP | No | None | -Colon adenocarcinoma (father) | MSH2 (c142 G>T) |
| Current report | M | 2 | 38 | Neck | MSH2/MSH6 | NP | Sebaceous adenomas | Urothelial carcinoma (41) | None | MSH2 (c.(?_68)_1276+?del) |
Abbreviations: KA: Keratoacanthoma; M: Male; F: Female; NR: Not reported; MMR: Mismatch repair protein expression; IHC: Immunohistochemistry.
A diagnostic algorithm (Fig. 2) for patients who are 50 years or younger and develop sporadic KAs is proposed. This algorithm rests on three pillars: the presence of a family history of KAs/colorectal cancer, immunohistochemical stains for MMR proteins and genetic studies of MMR genes and germinal alterations in the TGFBR1 gene.

We thank Dr Marta Pineda, Joan Brunet, Eleanor Reavey and David Goudie for assistance with the genetic study of the cases reported in this work and for comments that improved the manuscript.


