







El síndrome de Osler-Weber-Rendu, o síndrome hereditario hemorrágico telangiectasia, es un trastorno raro de herencia autosómica dominante con una prevalencia estimada de 1:10.000 personas a nivel mundial. Las manifestaciones clínicas de este síndrome son resultado de malformaciones arteriovenosas y varían desde telangiectasias en piel y mucosas hasta afección de órganos sólidos que ponen en peligro la vida, como alteraciones hepáticas, émbolos sistémicos y fallo cardíaco, por lo cual el diagnóstico oportuno es de suma importancia para prevenir las complicaciones de la enfermedad y proporcionar apoyo genético a los familiares. En esta revisión se analiza el cuadro clínico con enfoque principal en las manifestaciones mucocutáneas de la enfermedad y su abordaje terapéutico.
Osler-Weber-Rendu syndrome, also known as hereditary hemorrhagic telangiectasia, is a rare autosomal dominant disorder with an estimated worldwide prevalence of 1 case per 10,000 population. Its clinical manifestations are the result of arteriovenous malformations characterized by telangiectases that can affect the skin, mucous membranes, and solid organs and cause life-threatening conditions, such as liver disease, systemic emboli, and heart failure. Timely diagnosis is thus essential in order to prevent disease-related complications and offer genetic counseling to families. We review the clinical features of Osler-Weber-Rendu syndrome with a focus on mucocutaneous manifestations and their treatment.
El síndrome de Osler-Weber-Rendu, también conocido como síndrome hereditario hemorrágico telangiectasia (HHT), es un trastorno raro de herencia autosómica dominante en el cual existen comunicaciones anormales entre venas y arterias. Estas malformaciones arteriovenosas se manifiestan como telangiectasias en piel, mucosas y órganos sólidos1. La presencia de telangiectasias cutáneas representa un problema cosmético para los pacientes con HHT; sin embargo, son las manifestaciones sistémicas, como epistaxis recurrentes, alteraciones hepáticas, émbolos sistémicos y fallo cardíaco, las que ponen en riesgo su vida. La prevalencia estimada a nivel de mundial de HHT es de 1:5.000 y 10.000 personas, pero se considera que la enfermedad esta infradiagnosticada1,2. Los pacientes con HHT requieren un abordaje multidisciplinario debido a las diversas complicaciones sistémicas que pueden presentar. Es por ello que los médicos debemos estar familiarizados con este padecimiento y contribuir a un diagnóstico y tratamiento oportuno de la enfermedad.
HistoriaHistóricamente el primer reporte de HHT aparente fue hecho por Sutton3 en 1864 al describir un paciente masculino que presentaba episodios recurrentes de hemorragias y malformaciones vasculares. En 1865, Babington4 publicó un caso de epistaxis hereditaria en un niño y su madre que tenían episodios recurrentes de epistaxis. Posteriormente, en 1896, Rendu5 realizó la primera descripción del síndrome al identificar a un hombre con cuadro clínico de telangiectasias faciales en nariz, lengua y labios, quien además contaba con antecedente de anemia y episodios de epistaxis recurrentes desde la infancia y cuyo padre refería episodios de melena. Tiempo después, en 1901, Sir William Osler6 fue el primero en publicar la asociación familiar en el síndrome al describir a tres familias con telangiectasias y hemorragias hereditarias. En 1907 Weber7 describió con mayor detalle la asociación entre la herencia, telangiectasias y hemorragias. Fue en 1909 cuando Hanes8 propuso el nombre de hemorragia hereditaria telangiectasia.
EpidemiologíaEl síndrome HHT tiene una amplia distribución geográfica, y en los últimos años, debido a la mayor difusión y conocimiento de la enfermedad, los reportes de caso han ido en aumento, por lo que la prevalencia estimada es desconocida, con cifras que van desde 1:2.351 habitantes en Francia, 1:3.500 en Dinamarca y 1:39.216 en el norte de Inglaterra9. Se estima que a nivel mundial afecta a 1,4 millones de personas.
A pesar de que la mayoría de los pacientes tienen algún familiar con HHT, el 20% de los casos son esporádicos. Se estima que cada hijo de alguien con HHT tiene un 50% de riesgo de presentar la enfermedad10.
FisiopatologíaLas manifestaciones clínicas observadas en HHT son resultado de anormalidades en la estructura vascular. Las principales lesiones son las telangiectasias, que se producen por una comunicación arteriovenosa entre arteriolas y vénulas dilatadas y que clínicamente se presentan como máculas eritematosas puntiformes o lineales arborizantes de 1-2mm y que desaparecen a la presión leve11. El mecanismo fisiopatológico por el cual se producen las telangiectasias no está del todo dilucidado. La teoría de Braveman propone como mecanismo inicial una dilatación focal de vénulas poscapilares, las cuales continúan dilatándose progresivamente hasta conectarse con las arteriolas dilatadas a través de los capilares, los cuales posteriormente desaparecen y se forma una conexión arteriovenosa directa. Durante este proceso existe un infiltrado perivascular de predominio mononuclear12. Las telangiectasias por definición se presentan en tejidos mucocutáneos, como piel o mucosa gastrointestinal. Cuando estas comunicaciones vasculares anormales se producen en órganos internos, como hígado, pulmón y cerebro, se conocen como malformaciones arteriovenosas (MAV). Estas MAV son las responsables de las manifestaciones sistémicas de la enfermedad13. La fragilidad de las paredes vasculares y el flujo sanguíneo turbulento predisponen a estos vasos a causar las hemorragias características del síndrome14.
Se ha observado que el nivel del factor de crecimiento endotelial vascular (VEGF), que está involucrado en la vasculogénesis y la angiogénesis, se encuentra elevado en pacientes con HHT, por lo que se han desarrollado tratamientos contra este factor, como el bevacizumab, que han mostrado eficacia15.
A nivel molecular se han identificado más de 600 mutaciones causantes de HHT. De estas, las más estudiadas hasta la fecha son las mutaciones en el gen endoglina (ENG), localizado en el cromosoma9 característica del subtipo1 de HHT, y la del gen receptor de quinasa similar a activina (ALK1), en el cromosoma12 característica del subtipo 211,16. Ambas proteínas (ENG y ALK1) son proteínas transmembrana expresadas en las células endoteliales con capacidad de unión al factor de crecimiento transformante (TGF), el cual juega un papel importante en la angiogénesis y en la maduración vascular17. Además, se han descrito mutaciones en el gen MADH4 o SMAD4 que predisponen a HHT acompañada de poliposis juvenil11 (tabla 1). Otro tipo de mutación es la ocasionada en el receptor de la proteína morfogénica óseaII que produce el fenotipo HHT acompañado de hipertensión pulmonar primaria18.
Principales mutaciones en HHT
| ENG | Se asocia con mutaciones en endoglina.Más comúnmente observadas en MAV pulmonares y cerebrales |
| ACVRL1 | Se asocia con mutaciones en ALK-1 (receptor de quinasa similar a activina).Más comúnmente observadas en MAV hepáticas, hipertensión pulmonar y arterial |
| SMAD4 | Provoca poliposis juvenil asociada a HHT |
El cuadro clínico de HHT es muy variado, con manifestaciones principalmente en nariz, piel y pulmones, pudiendo llegar a afectar el sistema nervioso central y el gastrointestinal. Se caracteriza por la tríada clásica de epistaxis, telangiectasias y contar con el antecedente de un familiar afectado de HHT19.
La mayoría de los pacientes experimentan únicamente cuadros de epistaxis, telangiectasias mucocutáneas y anemia por deficiencia de hierro. Normalmente los síntomas no están presentes al nacimiento pero se desarrollan con la edad. Se estima que el 70% de los afectados presentan manifestaciones a los 16años y el 90%, a los 40años9.
NarizLa epistaxis suele ser la principal manifestación clínica, y en muchos de los casos la primera. Se produce por el sangrado espontáneo de las telangiectasias que están presentes en la mucosa nasal. Puede ser estimulada por múltiples factores, como cambios de temperatura, humedad o postura.
La frecuencia de los episodios de sangrado nasal puede ser tan alta que produzca anemia por deficiencia de hierro. Sin embargo, en otras ocasiones ocurren episodios únicamente de forma esporádica o incluso pueden llegar a no presentarse nunca, lo que hace la sospecha diagnóstica menos probable. Además, la epistaxis es considerada un síntoma muy común en la población general, por lo que lo vuelve poco sensible. Los episodios de sangrado nocturno son más específicos para el diagnóstico de HHT20.
Generalmente los episodios de epistaxis se inician en la infancia, alrededor de los 10años, y por lo común se vuelven más severos con el paso de los años. En promedio, los pacientes con HHT presentan alrededor de 18 episodios de epistaxis al mes. Rebeiz et al.21 desarrollaron una clasificación para la severidad de la epistaxis que está vigente hasta la fecha (tabla 2).
El manejo y la prevención de la epistaxis pueden variar desde intervenciones sencillas, como humidificación nasal, hasta medidas más agresivas, como cauterización, ablación con láser, cirugía, suplementación con estrógenos y embolectomía, dependiendo de la severidad del cuadro22,23.
PielLa principal manifestación cutánea son las telangiectasias, que ocurren hasta en el 75% de los pacientes; típicamente se presentan ya desde la infancia y aumentan en cantidad con la edad.
Las lesiones cutáneas suelen aparecer después del primer episodio de epistaxis. En promedio, los pacientes las visualizan entre la cuarta y la quinta décadas de la vida11.
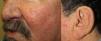
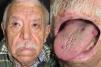
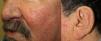
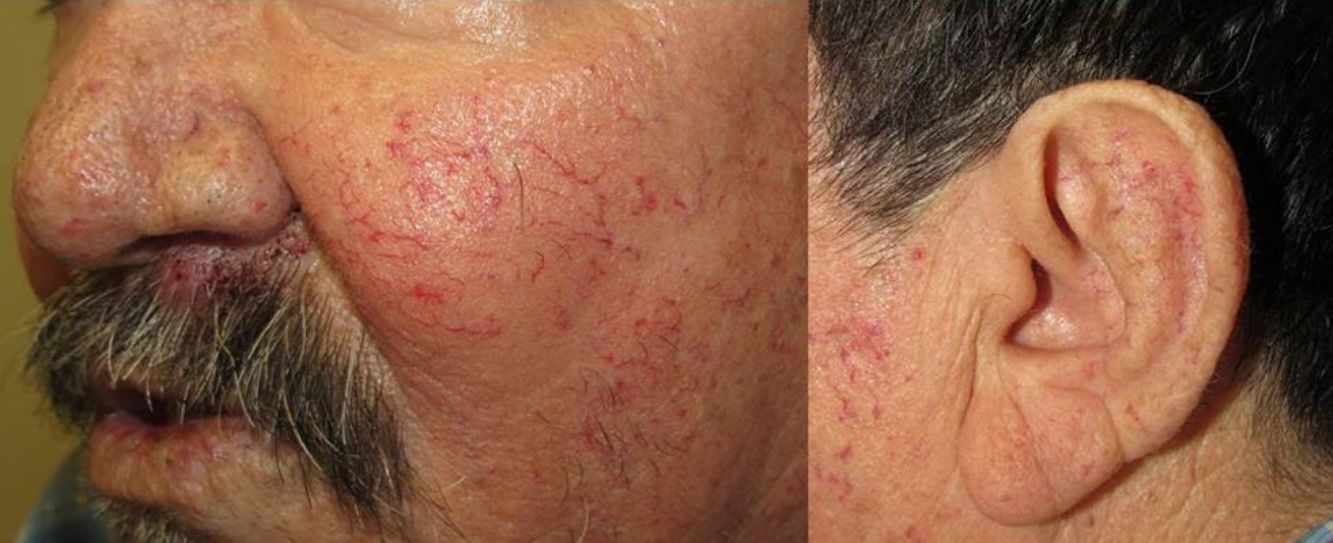
Los sitios donde se presentan con mayor frecuencia son la cara, los labios, la nariz, la lengua, los pabellones auriculares, las manos (especialmente en las puntas de los dedos), el tronco y los pies19 (figs. 1-3).
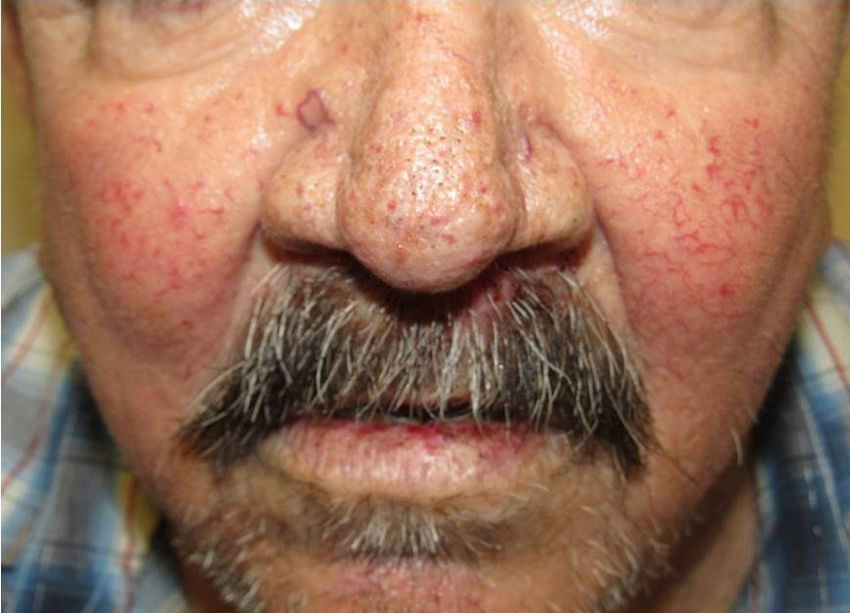
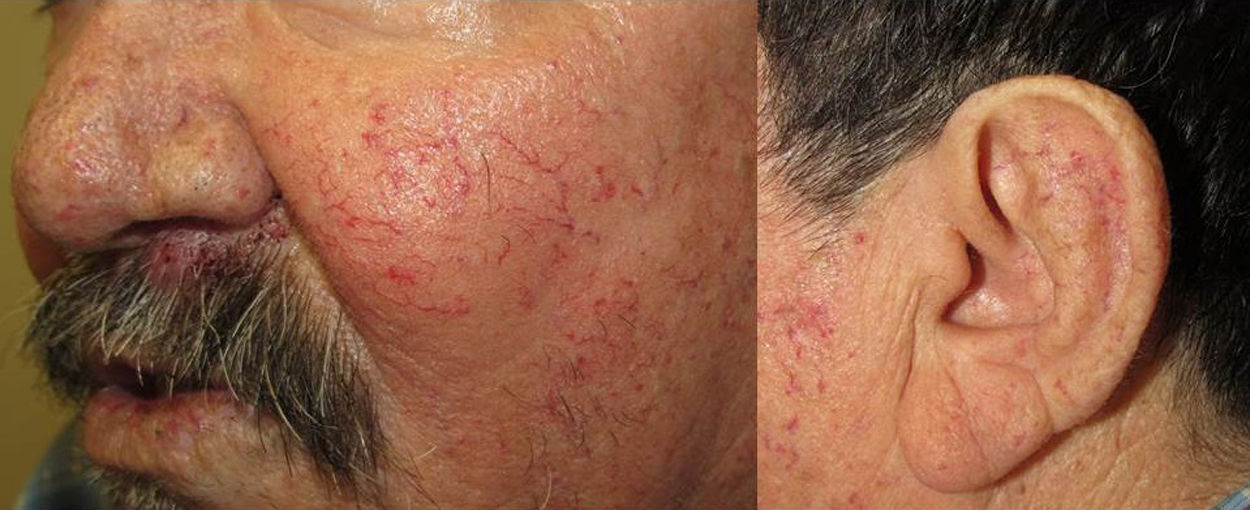
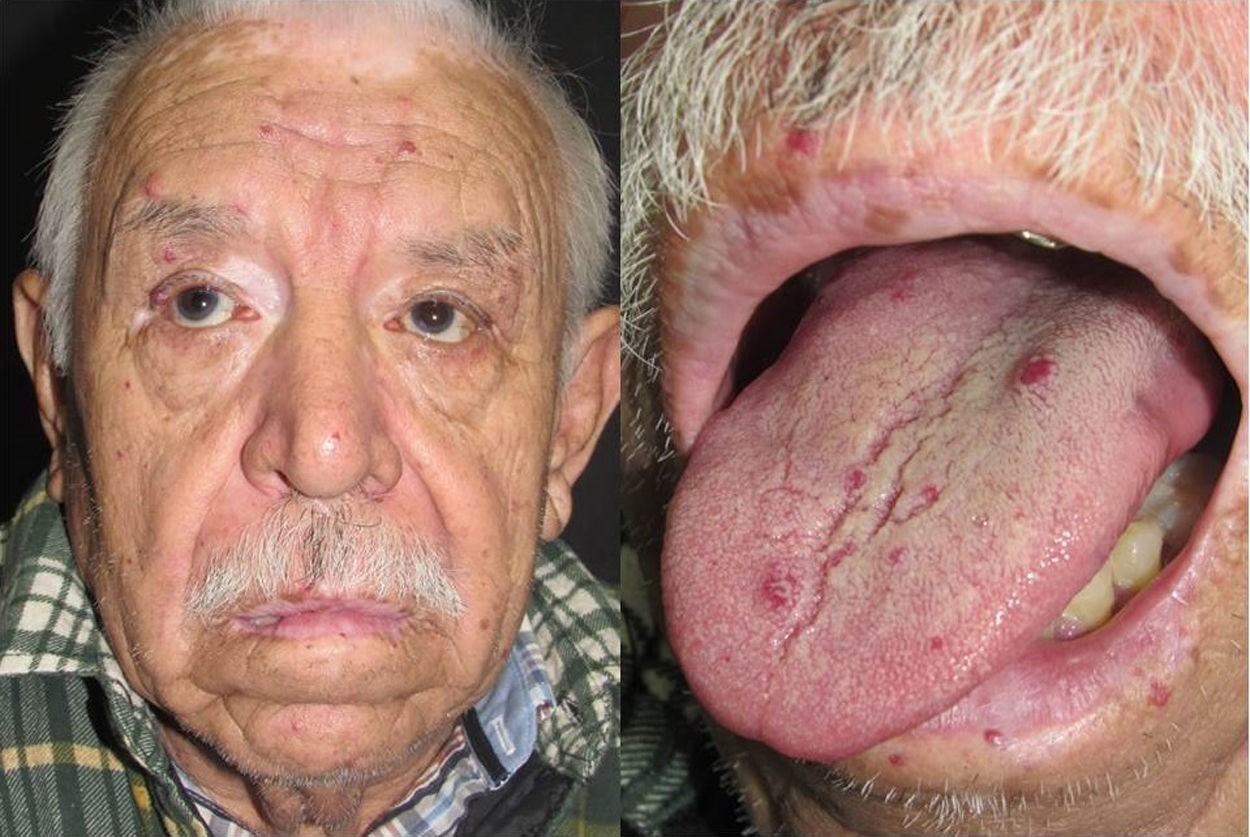
En la etapas iniciales se observan como máculas pequeñas rojo-violáceas de 1-3mm de diámetro, pulsátiles, y en la fase crónica pueden confluir y aumentar de tamaño formando lesiones arborizantes19,24.
En la mayoría de las ocasiones las telangiectasias cutáneas son asintomáticas y su tratamiento es únicamente por cuestión estética. Sin embargo, en algunos casos pueden ser dolorosas.
La Fundación Internacional de HHT realizó un consenso en el que se llegó a la conclusión de que se requiere por lo menos la presencia de 3 telangiectasias típicas para el diagnóstico de HHT17.
PulmónA nivel pulmonar las lesiones más frecuentes son las MAV, que se encuentran presentes hasta en el 15-30% de los casos de HHT. Estas MAV comunican la circulación pulmonar directamente con la sistémica, provocando un shunt de derecha a izquierda que se manifiesta con cuadros clínicos de cianosis, intolerancia al frío, migraña y policitemia. Entre sus complicaciones más severas se encuentran los accidentes cerebrovasculares, la hemoptisis masiva y el hemotórax espontáneo10.
Por lo general estas lesiones se encuentran en las bases pulmonares y sangran con mayor facilidad durante el embarazo25.
Debido a que las MAV pulmonares no producen síntomas en estadios tempranos y sus complicaciones pueden llegar a ser severas, se debe realizar una revisión a todos los pacientes con diagnóstico de HHT mayores de 16años por medio de una eco transtorácica. El tratamiento de estas lesiones se puede realizar con embolización transcatéter o, en casos más severos, con lobectomía10.
GastrointestinalA nivel gastrointestinal las manifestaciones del HHT se presentan en un tercio de los pacientes.
Las principales lesiones son telangiectasias en estómago y duodeno y posteriormente las MAV.
El cuadro clínico más común se presenta con anemia por deficiencia de hierro o episodios de sangrado gastrointestinal agudo; estas manifestaciones son más comunes a partir de los 40años10. La monitorización gastrointestinal se debe iniciar a los 35años y se realiza únicamente con medición anual de hemoglobina; la endoscopia se reserva para los episodios de sangrado. Para su tratamiento se puede utilizar ablación por endoscopia, embolización o cirugía.
CerebroLas lesiones en el cerebro se presentan hasta en el 23% de los pacientes. Estas pueden ser múltiples MAV, telangiectasias y fístulas arteriovenosas. Cuando se encuentran presentes se manifiestan como episodios migrañosos, convulsiones, isquemia o hemorragia. El rango anual de rotura de las MAV es del 2-4% en promedio. La realización de estudios de detección temprana es controvertida, pero si se decide realizar, la RM es el estudio de elección, con una sensibilidad de hasta el 80-95%. Para el tratamiento se puede realizar embolización o resección quirúrgica.
HígadoLa afección hepática en HHT se presenta en el 32-78% de los casos, siendo sintomáticas en el 8% de los ellos. La presentación clínica puede incluir angina y fallo cardiaco por el shunt que se produce entre la arteria y la vena hepática, que aumentan la sobrecarga cardíaca. También puede ocasionar hipertensión portal y encefalopatía hepática. Para la monitorización de estas lesiones se puede recurrir a la ecografía doppler, RM y TAC. El tratamiento es de soporte, con el objetivo de reducir las complicaciones de hipertensión portal y fallo cardíaco. En algunas ocasiones se puede recurrir al trasplante14.
DiagnósticoEl diagnóstico oportuno de HHT es de suma importancia para prevenir las complicaciones de la enfermedad y proporcionar apoyo genético a los familiares. En los casos en que se cumple la tríada clásica de epistaxis, telangiectasias y antecedentes familiares el diagnóstico es relativamente sencillo; sin embargo, el cuadro clásico no se presenta en todos los pacientes, por lo que en el año 2000 se publicaron los Criterios de Curaçao para el diagnóstico de HHT; en base a estos criterios, se requiere que estén presentes 3 o más para considerarlo como diagnóstico definitivo, 2 criterios positivos es igual a probable y 0 o ninguno, poco probable10,20 (tabla 3).
Existen también pruebas genéticas para el diagnóstico de HHT, las cuales son principalmente útiles en los familiares de pacientes afectados, especialmente niños y adultos jóvenes, que no cumplen los criterios diagnósticos clínicos. Dentro de estas pruebas se encuentra la secuenciación del ADN para detectar mutaciones en los genes ENG y ACVRL1, los cuales representan la mayoría de las mutaciones en HHT26. También existen pruebas para detectar mutaciones en el gen SMAD4, pero estas son menos comunes27.
CapilaroscopiaLa dermatoscopia puede convertirse en una herramienta para diagnóstico oportuno de HHT por medio de la capilaroscopia. Los hallazgos descritos hasta el momento sobre capilaroscopia en HHT muestran que es de utilidad, ya que permite detectar telangiectasias microscópicas. En un estudio realizado por Mager y Westermann17 usando microscopia capilar del pliegue ungueal encontraron que el 87% de los pacientes con HHT tienen alguna anormalidad vascular, siendo los vasos gigantes en forma de bucle el hallazgo más común.
TratamientoDebido a que el síndrome HHT tiene afección multisistémica, se requiere un tratamiento multidisciplinario. La mayoría de los estudios realizados sobre HHT son elaborados por otorrinolaringólogos debido a que la epistaxis es la principal afección de estos pacientes; sin embargo, a pesar de que las telangiectasias son igual de evidentes, existen pocos estudios sobre su tratamiento.
A pesar de que las telangiectasias cutáneas representan solamente un problema cosmético, su presencia puede afectar significativamente la calidad de vida de los pacientes, por lo que en el campo dermatológico se han implementado numerosas opciones de tratamiento con resultados variables. La mayoría de la información bibliográfica existente consiste únicamente en reportes de casos.
En los últimos años los tratamientos con láser han sido los más utilizados para el tratamiento de las telangiectasias cutáneas. Entre los más utilizados se encuentran el colorante pulsado (595nm), el cual es útil para la telangiectasias maculares, y el láser Nd:YAG, que se prefiere para las lesiones de aspecto papular por su mayor penetración y poder de coagulación28 (tabla 4).
Revisión de reportes de casos de telangiectasias en HHT tratadas con láser
| Estudio | Número de pacientes | Tratamiento | Zonas tratadas | Resultados |
|---|---|---|---|---|
| Cheung et al.28, 2015 | 1 | Láser colorante pulsado 595nmSpot 5mmDuración pulso 1,5msFluencia 8J/cm2 en mejillas y 9J/cm2 en nariz | Cara y mucosa | Resolución de las lesiones en 5días |
| Halachmi et al29., 2013 | 8 | Láser colorante pulsado (Vbeam Perfecta, Candela, Wayland)Spot 5-7mmDuración de pulso 1,5msFluencia 9,5 a 11J/cm2 | Mejillas, nariz y labios | Mejoría excelente en todos los pacientes (75-100%)Número promedio de sesiones para obtener respuesta: 2,6 |
| Werner et al.30, 2008 | 4 | Nd:YAGDuración de pulso: 0,5-90msFluencia máxima 450J/cm2Spot 3-10mm | Caso 1: mejillas, mentón y frenteCaso 2: punta de los dedosCaso 3: dedos, orejas, mejillas, mentónCaso 4: mejillas | Caso 1: 50-75% de respuesta a las 8semanasCaso 2: 75-95% de respuesta a las 8semanas; EA: pigmentación blanco-grisáceaCaso 3: 75-95% de mejoría en dedos después de 2sesiones; 50% de mejoría en orejas, mentón y mejillas con ligera atrofia e hipopigmentaciónCaso 4: respuesta 90% después de 8semanas |
| Fernández-Jorge et al.31, 2007 | 3 | Sistema Photoderm-Vasculight que combina IPL + Nd:YAG 1064nm | Mejillas, labios, nariz | Respuesta completa en todos los casos sin recurrencias en el seguimiento a 2años en 2 de los 3casos |
El láser de argón ha sido utilizado para el tratamiento de telangiectasias cutáneas; sin embargo, tiene la desventaja de producir cicatrices32.
Otros estudios han reportado el uso de luz pulsada intensa, seguida de láser Nd:YAG, con resultados cosméticos aceptables30.
ConclusionesEl síndrome de Osler-Weber-Rendu es una enfermedad multisistémica poco frecuente, con un amplio espectro clínico y un riesgo importante de desarrollar complicaciones, por lo cual requiere un diagnóstico y asesoramiento genético oportuno. Esta entidad no es completamente apreciada por los clínicos, quienes a menudo no reconocen la enfermedad hasta que se presentan complicaciones severas que comprometen la vida del individuo.
El pronóstico y la supervivencia de los pacientes con Osler-Weber-Rendu son favorables si las complicaciones son diagnosticadas y tratadas a tiempo.
El tratamiento con láser de las lesiones cutáneas ha mostrado ser prometedor, aunque se requieren más estudios a largo plazo para determinar con mayor exactitud su eficacia y estandarizar los parámetros necesarios según el sitio de la lesión a tratar.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


