



INTRODUCCION
Se conoce como síndrome de Bart a la combinación de ausencia localizada y congénita de piel, ampollas generalizadas y alteraciones ungueales 1. Tras unos años de seguimiento, varios de los miembros de las familias originales descritas por Bart seguían presentando ampollas durante la vida adulta. El estudio al microscopio electrónico de una biopsia de piel puso de manifiesto que las ampollas se formaban por debajo de la lámina densa y que las fibrillas de anclaje estaban malformadas. Por la clínica, el patrón de herencia y el estudio ultraestructural se llegó a la conclusión de que padecían una epidermólisis ampollosa distrófica dominante, como la mayoría de casos descritos posteriormente 2-4. Más recientemente, gracias a los estudios de mapas genéticos de la familia descrita por Bart, se ha comprobado que tienen una mutación en el cromosoma 3p, cerca del lugar de codificación del colágeno tipo VII (COL7A1) 5. La mutación consiste en una transición G-A en el exón 73, que da lugar a una sustitución glicina-arginina en el dominio triple hélice del colágeno tipo VII en los individuos afectados 6. El curso de la enfermedad ha sido favorable y el pronóstico excelente.
La epidermólisis ampollosa juncional tipo Herlitz (EAJ-H) es una enfermedad autosómico recesiva, rara y de muy mal pronóstico 7, que se caracteriza por fragilidad cutánea extrema y formación de ampollas generalizadas tras mínimos traumatismos. Los pacientes que la padecen suelen fallecer a lo largo del primer año de vida como consecuencia de una sepsis, facilitada por la ausencia de barrera cutánea, y distrés respiratorio. La EAJ-H está causada por mutaciones en los genes que codifican diferentes componentes de la laminina 5, filamento de anclaje que conecta los hemidesmosomas de los queratinocitos basales a la membrana basal subyacente 8,9. La mutación da lugar a hemidesmosomas hipoplásicos y escasos, lo que facilita la dehiscencia de la piel en la lámina lúcida.
Presentamos un caso de síndrome de Bart asociado a EAJ letal tipo Herlitz.
CASO CLINICO
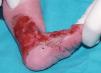
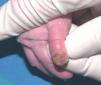
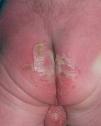
Se trata del segundo hijo varón nacido de padres sanos no consanguíneos, de nacionalidad marroquí. Presentaba desde el nacimiento una extensa área denudada por ausencia localizada y congénita de piel en la cara interna del tercio inferior del pie y el tobillo izquierdos (fig. 1), al que rodeaba por completo. Mostraba asimismo anoniquia, con lecho ungueal granulomatoso, en 3 dedos de las manos (fig. 2). Pocos días después desarrolló múltiples ampollas y erosiones sobre lugares de la piel expuestos a traumatismos como los dedos de manos y pies, pabellones auriculares, muslos, nalgas (fig. 3) y boca.
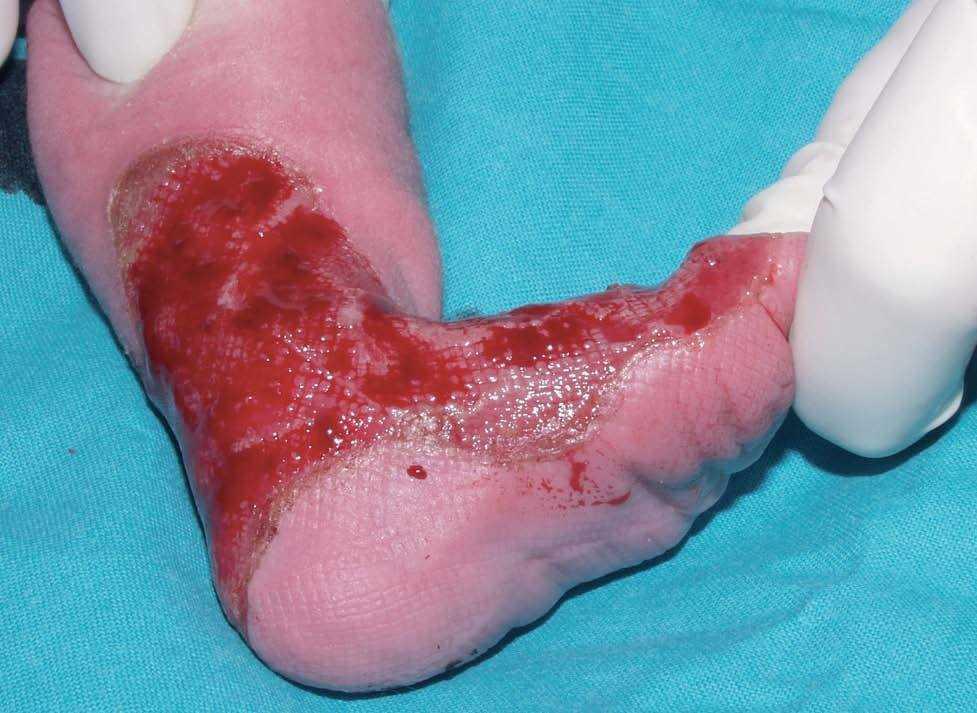
Fig. 1.--Ausencia localizada y congénita de piel.
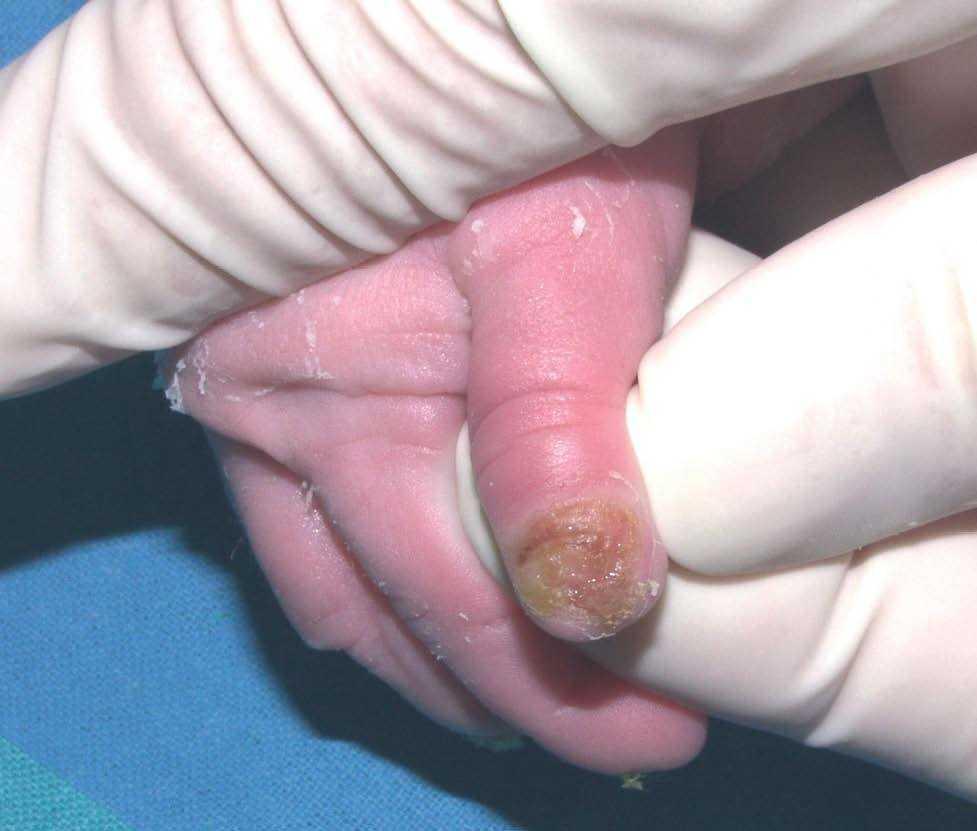
Fig. 2.--Anoniquia con lecho ungueal granulomatoso en el primer dedo.
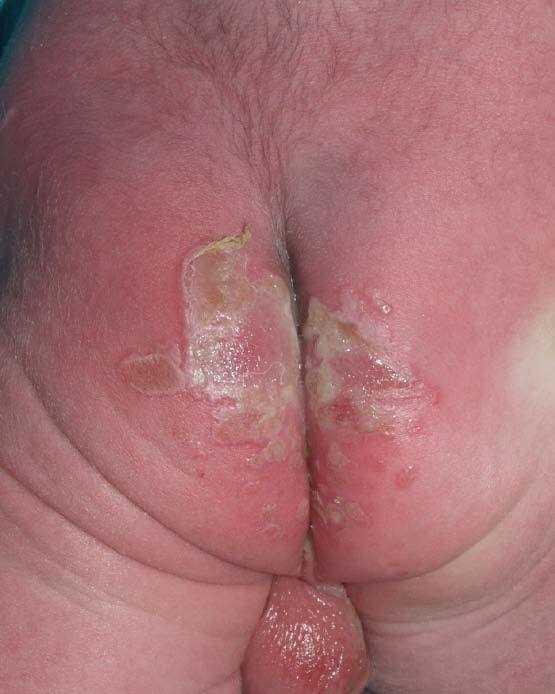
Fig. 3.--Múltiples erosiones y ampollas en las nalgas.
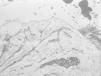
El estudio histológico de una biopsia cutánea con hematoxilina y eosina mostró separación subepidérmica sin infiltrados dérmicos. El estudio con microscopía electrónica evidenció ausencia de hemidesmosomas y rotura a través de la lámina lúcida (fig. 4).

Fig. 4.--Microscopio electrónico. Separación de la piel en la lámina lúcida. Hemidesmosomas hipoplásicos y escasos.
La inmunofluorescencia puso de manifiesto una ampolla subepidérmica. Los anticuerpos monoclonales contra las integrinas α5 y β4 y el BP180 tiñeron el techo de la ampolla. Los anticuerpos contra el colágeno IV (fig. 5) y colágeno VII tiñeron el suelo de la ampolla pero la tinción con anticuerpo GB3 (laminina 5) fue negativa.
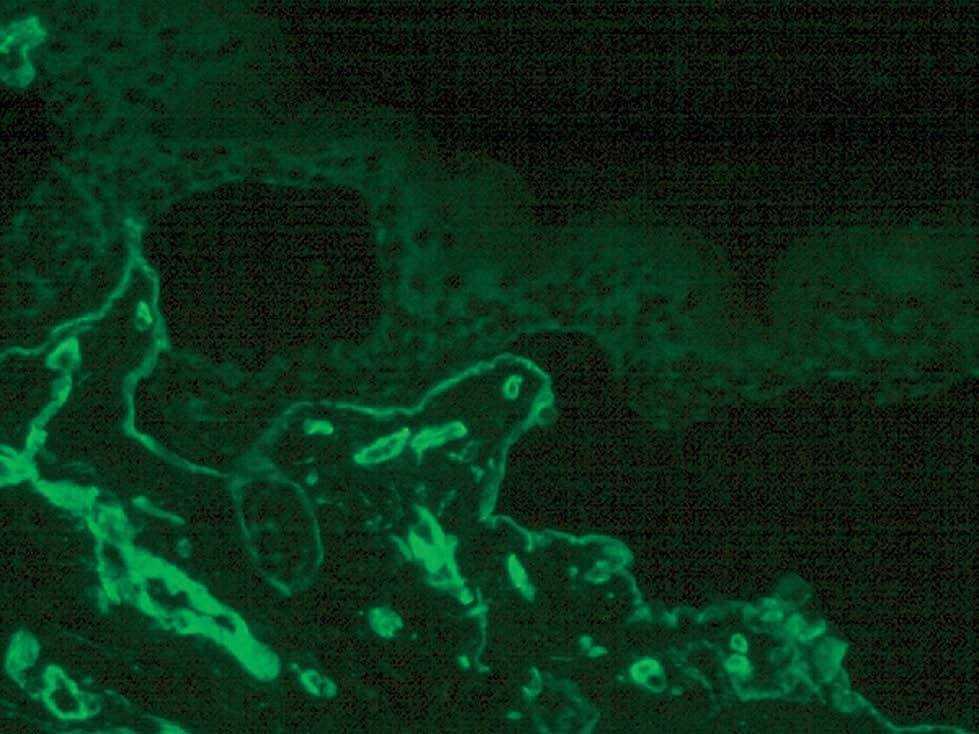
Fig. 5.--Inmunofluorescencia con anticuerpos anti-colágeno tipo IV en el suelo de la ampolla.
Con todo ello se estableció el diagnóstico de síndrome de Bart asociado a EAJ-H.
Durante las primeras semanas de vida la evolución se complicó con infecciones cutáneas y respiratorias recurrentes y se frenó el desarrollo pondoestatural, al rechazar el niño la ingesta de alimentos. El hemograma mostró una hemoglobina de 8,2 mg/dl y una albúmina sérica de 2,7 mg/dl. El resto de la bioquímica hemática fue normal.
En los meses sucesivos persistieron las erosiones cutáneas y mucosas, cada vez de mayor tamaño, y empeoró la anemia y el estado nutricional. El niño desarrolló finalmente una bronquiolitis con insuficiencia respiratoria y una sepsis grave que dio lugar a fallo cardíaco que condujo a la muerte del niño poco después de cumplir el séptimo mes de vida.
DISCUSION
Las EAJ forman un grupo de enfermedades ampollosas hereditarias en las que la separación dermoepidérmica se produce en la lámina lúcida de la región de la membrana basal. Clínicamente se han descrito 3 subtipos: a) la EAJ gravis, letal o tipo Herlitz (EAJ-H) (OMIM 226700), b) la EAJ atrófica generalizada (GABEB; OMIM 226650), una variante no letal con ampollas, atrofia cutánea, alopecia, distrofia ungueal e hipoplasia del esmalte dental y piqueteado dental, en la que se han identificado mutaciones en el gen COL17A1 10, y c) la EAJ con atresia de píloro.
La EAJ-H es una enfermedad autosómico recesiva, rara y habitualmente fatal 7. Su incidencia se sitúa alrededor de 1 caso por 200.000 habitantes. Se caracteriza por fragilidad cutánea extrema, con formación de ampollas de distribución generalizada en piel y mucosas, tras mínimos traumatismos, desde los primeros días de vida. Aparecen lesiones en las puntas de los dedos por el roce con las sábanas, y en la cintura o las extremidades por el mero hecho de sostener al niño entre los brazos. Las ampollas se rompen con facilidad dejando grandes erosiones cutáneas. A diferencia de las epidermólisis distróficas, las lesiones se resuelven sin dejar cicatriz ni quistes de millium. Suelen tardar en reepitelizar, lo que hace que se pierda la barrera cutánea durante periodos prolongados de tiempo, dejando al organismo sometido al riesgo de infecciones y de deshidratación durante largas temporadas. La afectación mucosa se produce principalmente en la boca, debido al roce con la tetina del biberón, y el tracto gastrointestinal. Se manifiesta como erosiones dolorosas, que dificultan la alimentación, y malabsorción, dando lugar a desnutrición grave. También se afecta la mucosa respiratoria, manifestándose al inicio con llanto ronco y más adelante con bronquiolitis. No hay uñas o son distróficas y habitualmente se observa un tejido de granulación exuberante periungueal. Posteriormente suele aparecer anemia y se enlentece el crecimiento. No es raro que la enfermedad se complique con sepsis y distrés respiratorio que contribuyen al pronóstico desfavorable. En la mayoría de casos la EAJ-H es letal muy precozmente, causando la muerte de la mayoría de los niños que la padecen a lo largo del primer año de vida 7.
La EAJ-H está causada por mutaciones en los genes que codifican diferentes componentes de la laminina 5, una molécula de adhesión de la matriz extracelular que descansa sobre la membrana basal y conecta ésta con los hemidesmosomas de la cara inferior de los queratinocitos basales 11. Se trata de una glucoproteína compuesta por 3 subunidades denominadas α3, β3, y γ2. Los genes de estas subunidades se localizan en el cromosoma 18q (3 LAMA3) y en el 1q25-32 (3 LAMB3 y 2 LAMC2). En caso de homocigosidad o heterocigosidad compleja esta glucoproteína no se expresa en la lámina lúcida y se detectan mutaciones nonsense, inserciones o deleciones 8,9 en ambos alelos de alguno de los 3 genes mencionados, aunque el que está alterado con más frecuencia es el gen LAMB3 (50 %). También se han descrito mutaciones hotspot como la R635X.
El diagnóstico de las epidermólisis ampollosas se confirma mediante el examen con microscopio electrónico de una biopsia cutánea de piel sana, donde se pueden observar los planos de rotura de la piel. En la EAJ-H la fisura se localiza en la lámina lúcida, donde pueden verse un número reducido de hemidesmosomas que además son hipoplásicos 12.
Una alternativa en el diagnóstico de las epidermólisis ampollosas es el mapeo antigénico por inmunofluorescencia, usando anticuerpos contra las proteínas estructurales de la región de la membrana basal tales como el antígeno del penfigoide ampolloso (BP180), la laminina y los colágenos tipo IV y VII. El conocimiento de la localización ultraestructural de dichos antígenos permite determinar el plano de hendidura. Como en nuestro caso, en la EAJ-H el mapeo antigénico muestra los colágenos tipo IV y VII en el suelo de la ampolla y el BP180 en la cara inferior del techo de la misma. Como es lógico, en la EAJ-H la tinción con anticuerpo antilaminina 5 (anticuerpo monoclonal GB3) es negativa o muy débil 12,13.
Para el diagnóstico prenatal puede usarse la biopsia fetal por fetoscopia y la biopsia amniótica. Ello ha permitido recientemente el aborto de un feto afectado y la progresión del embarazo de su gemelo sin mutación del gen LAMB 3, en un embarazo gemelar de una madre que previamente había tenido un hijo con EAJ-H 14.
En su artículo original, Bart describió una gran familia con una genodermatosis caracterizada por ausencia localizada y congénita de piel, junto con ampollas y alteraciones ungueales 1. Desde entonces se han descrito diversos pacientes con síndrome de Bart. Uno de ellos es un niño con epidermólisis ampollosa simple 15 en el que la microscopía electrónica y el inmunomapeo de las áreas con ausencia localizada de piel y de las zonas con fragilidad cutánea fueron idénticos. Pero en la mayoría de los casos, el síndrome de Bart se ha asociado a epidermólisis ampollosa distrófica dominante 2,3, tanto por sus características clínicas y ultraestructurales como inmunohistoquímicas y de herencia. Gracias al seguimiento se sabe que los pacientes que pertenecían a las familias originales descritas por Bart mostraban cicatrices atróficas y seguían presentando ampollas en la vida adulta. El análisis ultraestructural de la piel demostró la presencia de fibrillas de anclaje malformadas y formación de la fisura por debajo de la lámina densa 5. La tinción inmunohistoquímica localizó el colágeno tipo IV y VII en el techo de la ampolla. Los estudios de mapas genéticos de esta familia localizaron la mutación en el cromosoma 3p cerca del lugar de codificación del colágeno tipo VII (COL7A1). La mutación consistía en una transición G-A en el exón 73, que daba lugar a una sustitución glicina-arginina en el dominio triple hélice del colágeno tipo VII en los individuos afectados 6. Ninguno de los pacientes descritos había muerto como consecuencia de la enfermedad.
Nuestro paciente comenzó con ausencia localizada y congénita de piel desarrollando a los pocos días una EAJ-H. Sus características clínicas, formación de ampollas en piel y erosiones de la cavidad oral y uñas distróficas, la evolución fatal, la hendidura en la lámina lúcida y la falta de hemidesmosomas y de laminina en el estudio por inmunofluorescencia confirmaron el diagnóstico. Un paciente similar fue publicado por Skoven en 1979 16, aunque en dicho trabajo no se realizó el estudio antigénico ni la microscopía electrónica.
No estamos de acuerdo con Duran-McKinster et al 17 que sugieren que la ausencia localizada de piel en el síndrome de Bart sigue las líneas de Blaschko, por su bilateralidad y distribución simétrica en forma de banda ancha en forma de «S». En nuestro caso la banda denudada rodeó todo el tobillo, lo que hace difícil dicha explicación. Un trauma intraútero explicaría mejor esta morfología.
Nosotros creemos que el síndrome de Bart no es específico de un único subtipo de epidermólisis ampollosa, ya que se ha visto en todas las variantes de ésta: epidermolítica, juncional y distrófica.
Declaración de conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Josep Manel Casanova.
Servicio de Dermatología.
Hospital Universitari Arnau de Vilanova.
Universitat de Lleida.
Rovira Roure, 80. 25199 Lleida. España.
jmcasanova@medicina.udl.es
Recibido el 12 de febrero de 2006.
Aceptado el 26 de mayo de 2006.


