


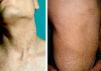
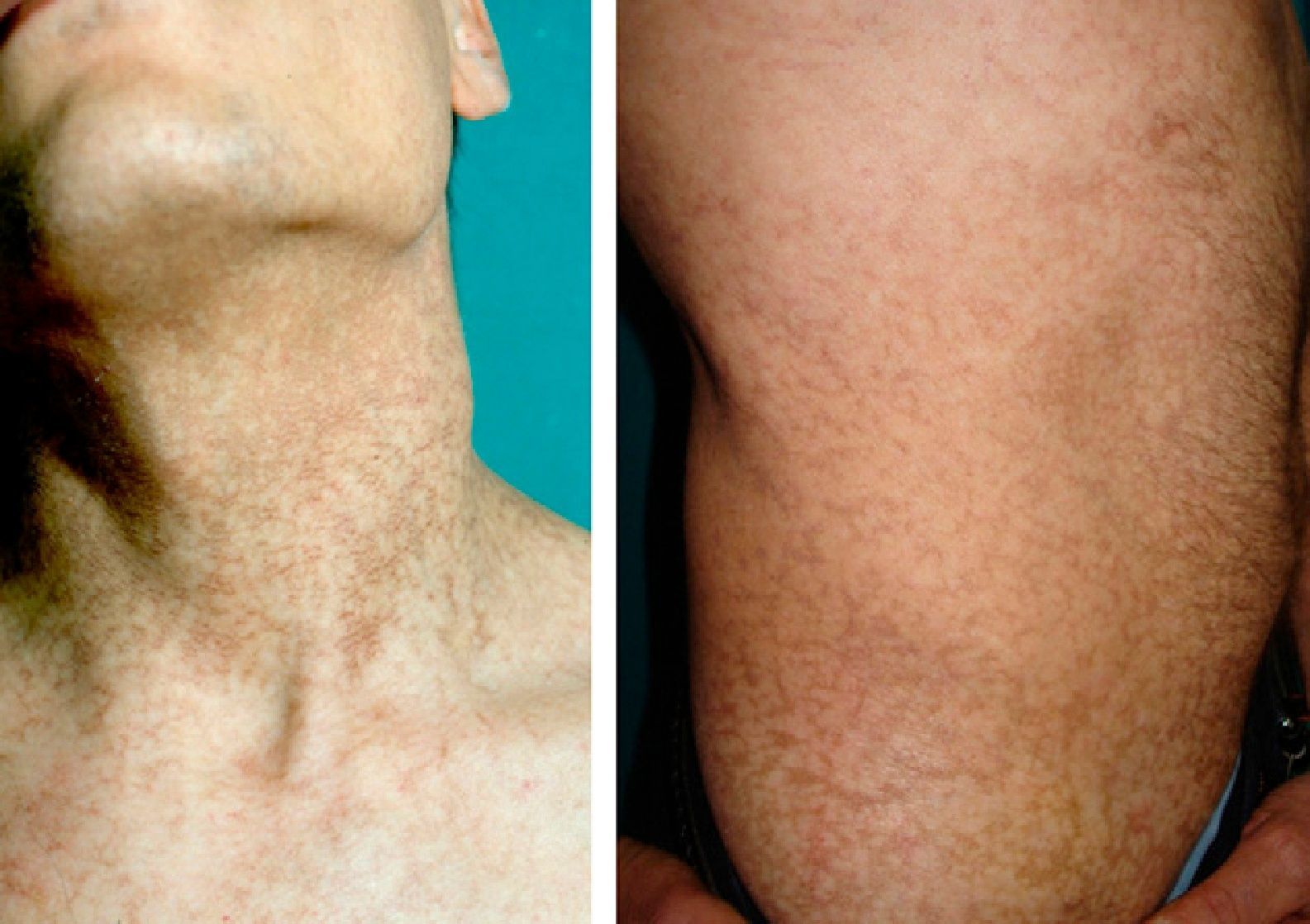
A man and his son had been examined at our dermatology department 19 years earlier for reticulate hyperpigmentation of the neck. The father had not attended subsequent follow-ups but was recently referred to our clinic by the hematology department, where he was being followed for neutropenia and thrombopenia. His son had died of severe aplastic anemia.
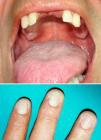
Physical ExaminationWhen the patient was 28 years old, he developed reticulate hyperpigmentation of the neck and upper trunk, with palmoplantar hyperkeratosis and hyperhidrosis and nail dystrophy (Fig. 1). His son (aged 8 years at the time) also began to experience similar pigmentary changes and nail dystrophy.
At the time of the current referral (the patient is now 47 years old), the reticulate pigmentation had spread, numerous teeth had been lost, and a small patch of leukoplakia had appeared on the buccal mucosa (Fig. 2).
Histopathology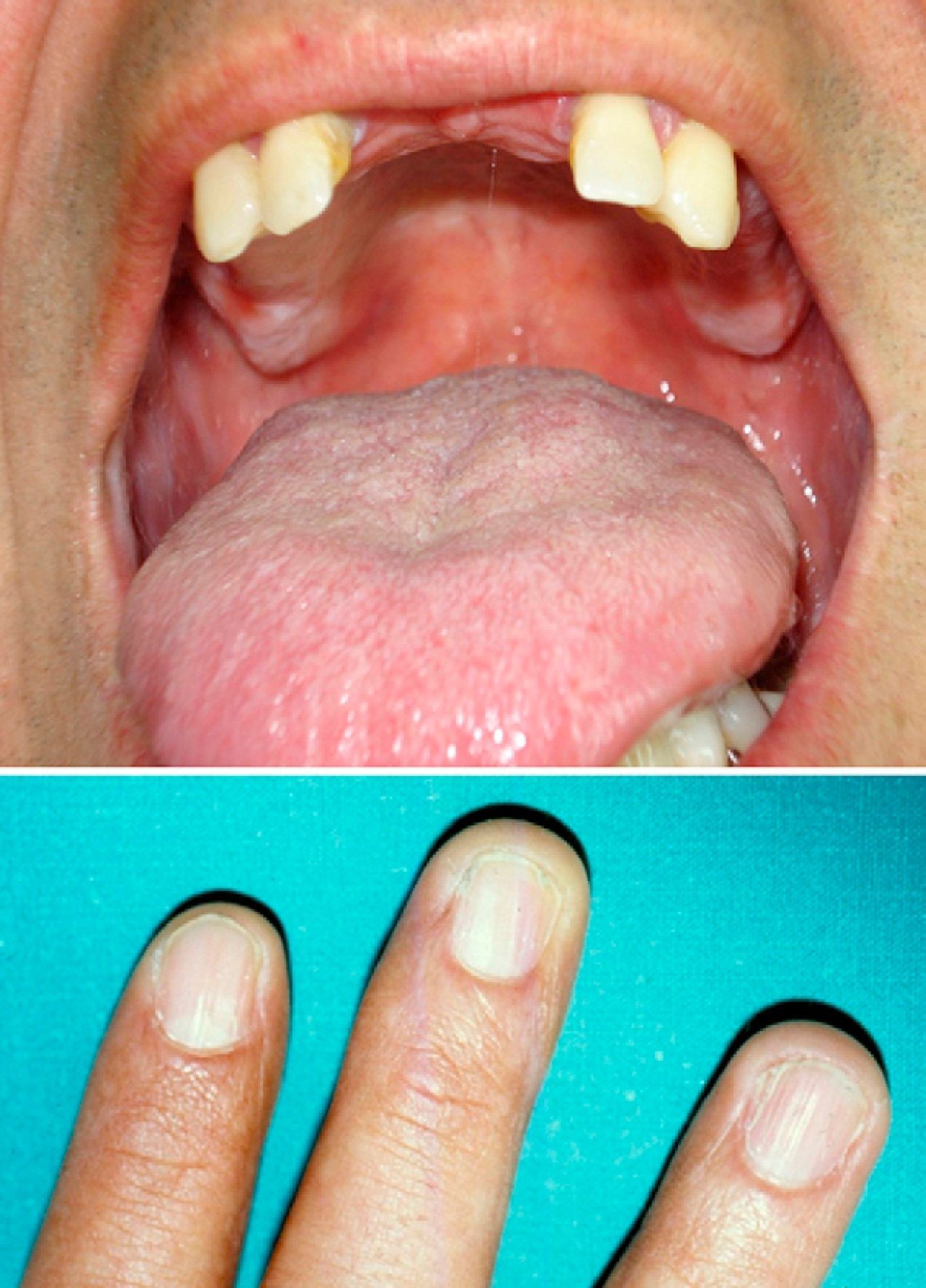
A mucosal biopsy showed a thinned epidermis with melanophages in the superficial dermis.
Additional TestsBlood tests showed neutropenia and thrombopenia. A bone-marrow biopsy revealed grade 2 aplastic anemia with loss of the megakaryocytic series and a reduction in the number of granulocytes. A peripheral blood karyotype was normal and mitomycin C did not induce chromosome breakage.
What Is Your Diagnosis?
DiagnosisDyskeratosis congenita.
Clinical Course and TreatmentPeriodic follow-up visits with a dermatologist were begun again so that malignant growths could be detected early. The hematology department closely monitored the patient's hematologic disorders and introduced treatment with thrombopoiesis-stimulating factors.
CommentDyskeratosis congenita, also known as Zinsser-Engman-Cole syndrome, is a genodermatosis with severe multisystem complications characterized by reticulate skin pigmentation, nail dystrophy, and leukoplakia.1 Telomere maintenance molecule defects are present. The underlying genetic abnormality is heterogeneous, and several mutations of the telomerase complex have been described.2,3 X-linked recessive, autosomal dominant, and autosomal recessive inheritance patterns have been observed; the first of the three is the most common. In the case we describe, the information available (male patient whose son had the syndrome even though he was not the offspring of a consanguineous relationship) suggest autosomal dominant transmission. This inheritance pattern has been linked to anticipation, whereby symptoms appear earlier and are more severe in successive generations, in relation to progressive telomere shortening.4
The prognosis of patients with dyskeratosis congenita is poor. Bone marrow failure (which occurs in up to 50% of cases) and a predisposition to malignant neoplasms (especially epidermoid carcinomas in areas of leukoplakia) are the main causes of early death in these patients.5 Among the many other clinical findings that have also been described are palmoplantar hyperkeratosis, hyperhidrosis, premature graying of hair, epiphora, caries and tooth loss, mental retardation, short stature, lung involvement, and liver fibrosis.1,2,5
Fanconi anemia, Naegeli-Franceschetti-Jadassohn syndrome, and dermatopathia pigmentosa reticularis must be considered in the differential diagnosis.6 Fanconi anemia also manifests with pancytopenia, pigmentary disorders, and predisposition to malignancies. It is, however, a more diffuse hypermelanosis associated with bone abnormalities and chromosome breakage induced by mitomycin C. Naegeli-Franceschetti-Jadassohn syndrome has no leukoplakia or bone marrow involvement and reticulate hyperpigmentation disappears in adolescence. Although dermatopathia pigmentosa reticularis also involves hyperpigmentation and onychodystrophy, it is characterized by the presence of nonscarring alopecia and absence of bone-marrow involvement.
An interdisciplinary approach is recommended to treat the complications these patients may develop. Close follow-up by a dermatologist is required for early detection of epidermoid carcinomas in areas of leukoplakia.6
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Armengot-Carbó M, et al. Hiperpigmentación reticulada y aplasia medular. Actas Dermosifiliogr. 2013;104:249–50.


