


El síndrome de Olmsted es un trastorno de la queratinización poco frecuente. Se caracteriza por una queratodermia palmoplantar mutilante y bilateral, que asocia la presencia de placas queratósicas localizadas alrededor de la boca, de la nariz y del ano. Además, a menudo presentará una deformidad en flexión de los dedos y anomalías ungueales1. En el presente artículo se describe el caso clínico de una niña con síndrome de Olmsted, la cual presentaba placas queratósicas, de tipo pitiriasis amiantácea, en el cuero cabelludo e hiperqueratosis folicular en las piernas.
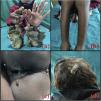
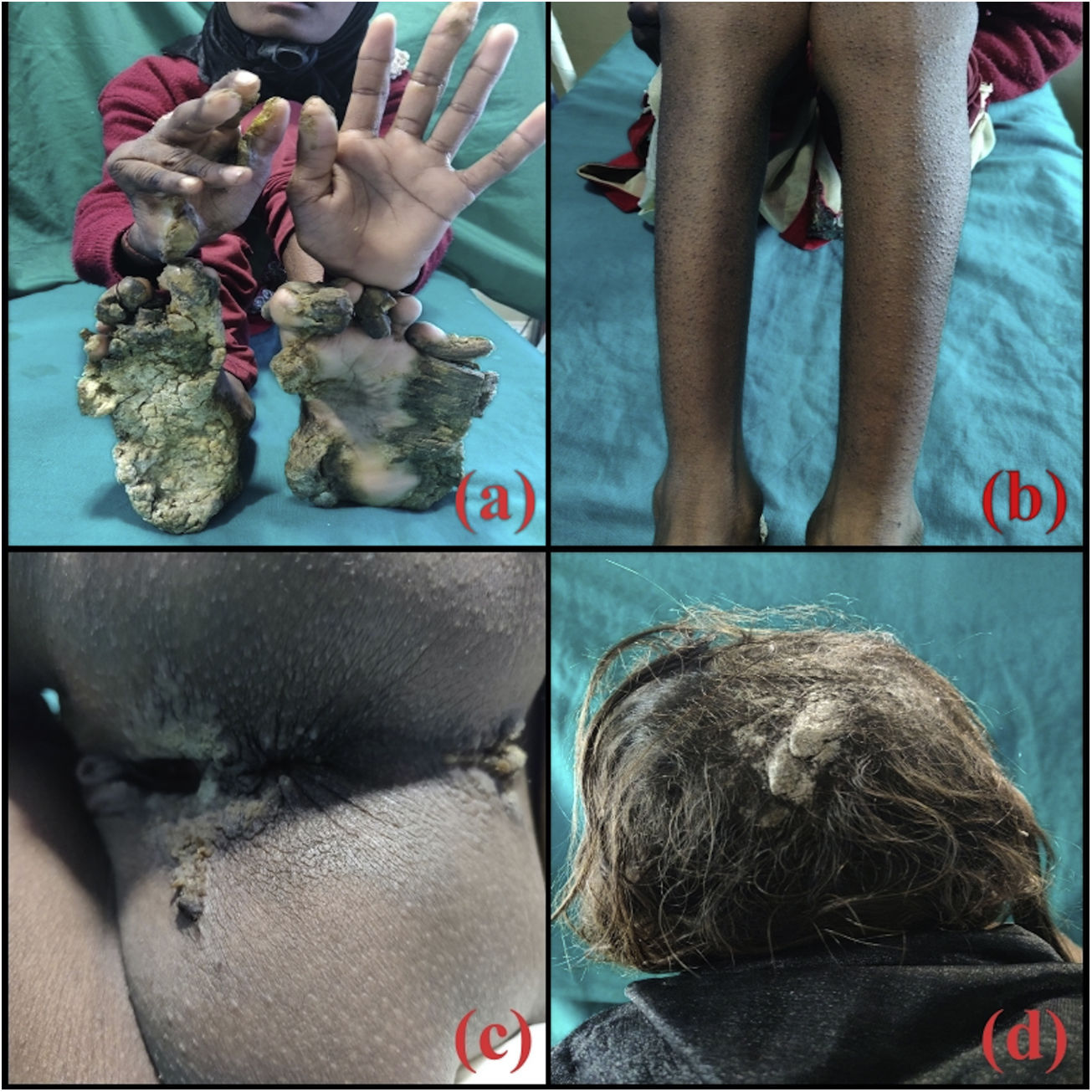
Una niña de 10 años consulta por placas hiperqueratósicas simétricas, bien delimitadas, localizadas en las palmas y en las plantas. Las placas aparecieron cuando la niña tenía 2 meses. Las lesiones eran dolorosas, por lo que cuando estaba descalza caminaba con cierta dificultad. Como antecedentes personales de importancia los padres referían nacimiento por parto eutócico y negaban antecedentes de consanguinidad. Incluyendo a los 2 hermanos, negaban antecedentes familiares de lesiones cutáneas similares. El desarrollo psicomotor estaba dentro de los parámetros normales. En la exploración se objetivaba una niña activa, sin ningún hallazgo significativo. La exploración cutánea reveló la presencia de placas hiperqueratósicas localizadas en las palmas y en las plantas, dando una imagen de «sandalias queratósicas» (fig. 1a). Además, la niña presentaba pápulas hiperqueratósicas perifoliculares localizadas en ambos miembros inferiores (fig. 1b). Por otro lado, tenía una placa hiperqueratósica y verrucosa localizada en la hendidura interglútea (fig. 1c); sin embargo, la región perioral estaba conservada. La niña presentaba también una deformidad en la flexión de la articulación metacarpofalángica de la mano derecha, de aproximadamente 5 años de evolución, además de onicogrifosis en las 3 primeras uñas de ambos pies y onicodistrofia de las uñas de ambas manos. En los últimos 4 años había presentado una disminución tanto en el grosor como en el número de pelos de las cejas y de las pestañas. Por otro lado, se observaban placas hiperqueratósicas localizadas predominantemente en la región occipital del cuero cabelludo (fig. 1d). Finalmente, no se evidenciaban hallazgos patológicos en las mucosas bucales, genitales, oculares o dentales; así mismo, las articulaciones se encontraban dentro de la normalidad.
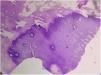
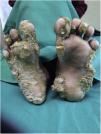
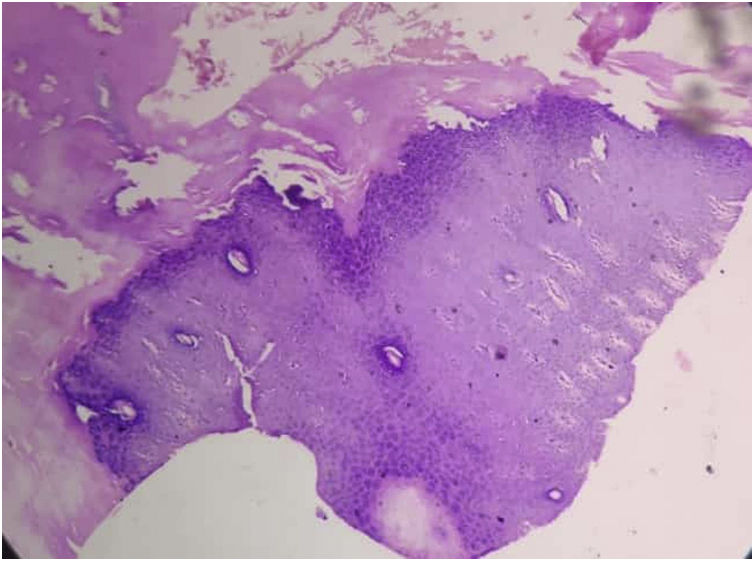
Tanto las pruebas rutinarias de laboratorio como el nivel sérico de cinc estaban dentro de los parámetros normales. La radiografía de las manos demostró la presencia de una deformidad en flexión de la mano derecha. El examen histopatológico mostró una amplia hiperqueratosis, asociada a paraqueratosis, hipergranulosis y acantosis (fig. 2). Se decidió iniciar tratamiento con acitretina en cápsulas (0,5mg/kg/día) en combinación con urea tópica y ácido salicílico. Tras 8 semanas de tratamiento se observó la mejoría parcial de las lesiones (fig. 3); sin embargo, la paciente se perdió durante el periodo de seguimiento.
Olmsted describió por primera vez esta enfermedad en un niño de 5 años, quien presentaba una queratodermia palmoplantar bilateral y simétrica, con bordes bien delimitados; esta clínica se asociaba además a placas hiperqueratósicas en la región periorificial, presentes desde el primer año de vida. La mayoría de los casos de este síndrome suelen ser esporádicos, aunque también se pueden encontrar formas autosómicas dominantes, autosómicas recesivas y ligadas al cromosoma X. La mutación de ganancia de función en el potencial de receptor transitorio vaniloide-3 ?TRPV-3? en el cromosoma 17 es la más común en los pacientes con el síndrome de Olmsted, alteración que conllevará un incremento de la apoptosis de los queratinocitos1. En las variantes recesivas y ligadas al cromosoma X se ha descrito la presencia de una mutación en el gen MBTPS-2 que codifica para una metaloproteasa del cinc. Los estudios inmunológicos han demostrado una proliferación de queratinocitos basales y suprabasales con reactividad para el marcador K672. La afectación en los varones será más frecuente que en las pacientes de sexo femenino3.
El cuadro clínico generalmente se evidencia cuando el niño empieza a andar y a coger objetos. La queratodermia palmoplantar es de tipo transgrediens y será gravemente incapacitante. Las pápulas queratósicas se desarrollan en áreas periorificiales y pueden extenderse a sitios de flexuras. Se producirán fisuras en los dedos de los pies, que conducirán a la autoamputación de los mismos, y el engrosamiento cutáneo puede provocar deformidades en flexión4. Nuestra paciente presentaba una queratodermia mutilante, pápulas periorificiales y deformidad en flexión. Sin embargo, no hubo formación de «pseudoainhum». Además, la paciente presentaba pápulas foliculares queratósicas distribuidas por ambas piernas.
Los pacientes con síndrome de Olmsted se caracterizan por presentar anomalías capilares tales como alopecia, cabello escaso, fino, lanoso y rizado, así como madarosis5. Nuestra paciente tenía una pérdida difusa del cabello, así como cabello escaso, fino y de color castaño. Además, en el cuero cabelludo tenía unas placas secas y gruesas, las cuales clínicamente se asemejaban a una placa de pitiriasis amiantácea. Estos pacientes pueden tener uñas distróficas, sin brillo, rugosas y ásperas; además, pueden presentar onicogrifosis, onicolisis, hiperqueratosis subungueal e incluso pueden carecer de uñas5. La baja estatura, característica en el síndrome de Olmsted, se deberá a un retraso en el desarrollo físico. Otras características clínicas incluyen la leucoqueratosis oral, la hiperhidrosis palmoplantar, la hipohidrosis y la presencia de opacidades corneales, de anomalías dentales y de estrías hiperqueratósicas localizadas en los pliegues cutáneos. Se ha descrito el desarrollo de carcinomas de células escamosas y de melanomas cutáneos a partir de las lesiones palmoplantares6.
La presencia de placas queratósicas periorificiales en un paciente con queratodermia palmoplantar será diagnóstica del síndrome de Olmsted y ayudará a excluir otras causas de queratodermia palmoplantar, como son el mal de Meleda, la paquioniquia congénita y el síndrome de Vohwinkel. La mutación del gen KDSR, que codifica para la enzima 3-cetodihidroesfingosina reductasa, involucrada en la síntesis de las ceramidas, también puede presentarse con características clínicas similares al síndrome de Olmsted. Sin embargo, estos pacientes tienen una queratodermia palmoplantar más leve, con gran eritema y placas descamativas bien delimitadas localizadas en la cara y en los genitales7.
El tratamiento del síndrome de Olmsted es difícil. La queratodermia puede controlarse con la combinación de agentes queratolíticos (ácido salicílico, urea, ácido bórico, aceite de esquisto), emolientes y retinoides orales. El uso de vendajes húmedos, así como la inmersión prolongada en agua de la parte afectada, se ha intentado utilizar con resultados variables8. Se ha probado con diversos tratamientos sistémicos, como es el caso de los antihistamínicos, de la vitamina E y la vitamina A, de los antimicrobianos y corticosteroides; sin embargo, no se han obtenido grandes beneficios. Tras pautarse acitretina (0,5mg/kg/día) asociada a un queratolítico tópico (20% de urea) y a emolientes, nuestra paciente obtuvo un alivio parcial. En los pacientes resistentes al tratamiento se puede intentar la extirpación quirúrgica seguida de un injerto de piel. En una serie de casos de 3 pacientes recientemente publicada, el erlotinib, un inhibidor del factor de crecimiento epidérmico, fue descrito como una posibilidad terapéutica para mejorar la hiperqueratosis y el dolor9.
Hasta este momento, en la literatura se han reportado menos de 80 casos de síndrome de Olmsted. Nuestra paciente es un caso único, ya que presentaba una descamación gruesa localizada en el cuero cabelludo, así como pápulas queratósicas en las piernas, además de las manifestaciones clínicas habituales observadas en el síndrome de Olmsted, como son la queratodermia palmoplantar mutilante, la deformidad en flexión y la presencia de hiperqueratosis en la hendidura interglútea.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


