





Morphea or localized scleroderma is a distinctive inflammatory disease that leads to sclerosis of the skin and subcutaneous tissues. It comprises a number of subtypes differentiated according to their clinical presentation and the structure of the skin and underlying tissues involved in the fibrotic process. However, classification is difficult because the boundaries between the different types of morphea are blurred and different entities frequently overlap. The main subtypes are plaque morphea, linear scleroderma, generalized morphea, and pansclerotic morphea. With certain exceptions, the disorder does not have serious systemic repercussions, but it can cause considerable morbidity. In the case of lesions affecting the head, neurological and ocular complications may occur. There is no really effective and universal treatment so it is important to make a correct assessment of the extent and severity of the disease before deciding on a treatment approach.
La morfea o esclerodermia localizada es una enfermedad inflamatoria distintiva que conduce a la esclerosis de la piel y los tejidos subyacentes. Incluye una serie de entidades que pueden distinguirse en base a las manifestaciones clínicas y la estructura de la piel y los tejidos subyacentes involucrados en el proceso fibroso. Sin embargo, la clasificación de estos procesos resulta difícil desde el momento en que los límites entre ellos no siempre son claros y es frecuente el solapamiento. En esencia, se distingue entre la morfea en placas, la esclerodermia lineal, la morfea generalizada y la panesclerótica. Si bien no tiene, salvo excepciones, una repercusión sistémica grave, sí que puede ser causa de una gran morbilidad. Si las lesiones asientan en el polo cefálico, pueden acompañarse de complicaciones neurológicas y oculares. No existe un tratamiento realmente eficaz y universal por lo que es importante realizar una evaluación correcta de la extensión y la gravedad de la enfermedad antes de tomar una decisión terapéutica.
The synonymous terms morphea and localized scleroderma refer to a distinctive inflammatory disease that primarily affects the skin and underlying tissues and ultimately leads to sclerosis. Localized scleroderma is differentiated from the systemic form of the disease by the absence of sclerodactily, Raynaud phenomenon, abnormalities of the nail-bed capillaries, or internal organ involvement.1 It is a rare disease, with an incidence that varies between 0.34 and 2.7 cases per 100 000 population per year.2,3 It is more common in white women (female to male ratio between 2.4:1 and 4.2:1)3–6 and its prevalence is similar in children and adults.6
ClassificationLocalized scleroderma includes a number of conditions that can be distinguished by their clinical manifestations and by the structure of the skin and subcutaneous tissue affected by the fibrosis. However, classification can be difficult because the boundaries between the different types are not always clear, and overlap is common. In 1995, after reviewing the medical literature, Peterson et al.7 proposed a classification of localized scleroderma that has been adopted with few modifications in the majority of review articles published since that time.8–14 In their classification, Peterson et al. used the term morphea consistently for all clinical types, describing plaque morphea, generalized morphea, bullous morphea, linear morphea, and deep morphea. They also defined different subtypes within each of these groups (Table 1). In addition, those authors proposed the inclusion of a number of diseases whose relationship with morphea had been suggested previously but which were, and in some cases still are, a source of controversy; for example, they added atrophoderma of Pasini and Pierini and lichen sclerosus et atrophicus to the plaque morphea group, progressive facial hemiatrophy to the linear morphea group, and eosinophilic fasciitis to the deep morphea group. Finally, they introduced the idea that this classification was not restrictive, meaning that the different categories were not mutually exclusive and that different subtypes could often be observed in a single patient.
Classification of Morphea or Localized Scleroderma.
| Plaque morphea |
| Plaque morphea |
| Guttate morphea |
| Atrophoderma of Pasini and Pierini |
| Keloid morphea (nodular morphea) |
| (Lichen sclerosus et atrophicus) |
| Generalized morphea |
| Bullous morphea |
| Linear morphea |
| Linear morphea (linear scleroderma) |
| Morphea en coup de sabre |
| Progressive facial hemiatrophy |
| Deep morphea |
| Morphea profunda |
| Subcutaneous morphea |
| Eosinophilic fasciitis |
| Pansclerotic morphea of childhood |
Source: Peterson et al.7
In 2004, a working group of the Paediatric Rheumatology European Society15 published a new proposal for the classification of juvenile localized scleroderma with the aim, in their opinion, of correcting certain deficiencies in the classification put forward by Peterson et al. They excluded atrophoderma of Pasini and Pierini, lichen sclerosus et atrophicus, and eosinophilic fasciitis, and proposed minor modifications to the organization of the different subtypes. The essence of the previous classification was maintained and the concept of mixed morphea was added to make allowance for patients presenting a combination of 2 or more types of lesion (Table 2). In 2006, the same working group published a study that included a large number of children with localized scleroderma and highlighted the need to incorporate the concept of mixed morphea, as up to 15% of the children presented a combination of lesions of different forms of morphea (Fig. 1).5 This same approach can be used in adults. In reality, recognition of this subgroup is implicit in the classification by Peterson et al., as they accepted that a single patient can develop different subtypes of morphea simultaneously.
Classification of Morphea or Localized Scleroderma.
| Circumscribed morphea |
| a) Superficial |
| b) Deep |
| Linear scleroderma |
| a) Trunk/limbs |
| b) Head |
| Generalized morphea |
| Pansclerotic morphea |
| Mixed morphea |
Source: Consensus Conference, Padua (Italy), 2004.15
Plaque morphea is the most superficial form, as the fibrotic process is often limited to the dermis, whereas the fibrosis in linear morphea affects not only the dermis but also the adipose tissue, muscle, and in many cases even bone. In deep morphea, the fibrosis affects the deep dermis, adipose tissue and muscle but, in contrast to the linear form, the lesions are more diffuse and do not follow a linear pattern.
Although serious systemic repercussions only develop in very rare cases of localized scleroderma, the disease can be a source of considerable morbidity as the lesions often affect the face and limbs and can lead to marked deformity and significant physical disability. In general, the deeper and more extensive the sclerotic process, the greater the likelihood of some associated visceral abnormality. This visceral involvement occurs principally in linear scleroderma, generalized morphea, and deep morphea. The most common systemic complications are joint pain and, when lesions affect the head, neurological and ocular manifestations. Relatives of patients with morphea are also at higher risk of developing autoimmune diseases.5,6
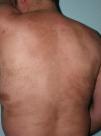
Clinical variantsPlaque (Circumscribed) MorpheaPlaque morphea, also known as circumscribed morphea, is the most common variant of localized scleroderma in the adult.3,4,6 The sclerotic changes mainly affect the reticular dermis. This condition presents as clearly circumscribed, oval or round areas of shiny indurated skin in 1 or at most 2 anatomical regions, most frequently the trunk or limbs (Fig. 2). In the earliest phases there is a highly characteristic, violaceous halo around the plaque; this represents the most inflammatory phase of morphea. Atrophoderma of Pasini and Pierini is the term used when lesions develop from the outset as slightly depressed plaques of grayish-brown color. These lesions often affect the trunk or the proximal areas of the limbs.9,13 Most authors agree that the lesions of atrophoderma of Pasini and Pierini, in which the skin is not indurated, constitute a variant of morphea, either an abortive form of the disease9,16 or an even more superficial variant in which the sclerotic changes affect only the papillary or superficial dermis.17 There is evidence to support the relationship between these lesions and morphea. First, the lesions of atrophoderma of Pasini and Pierini coexist with the typical lesions of plaque morphea in 20% of cases16 and, secondly, microscopy in plaque morphea has shown that sclerosis limited to the most superficial layers of the reticular dermis is observed clinically as thinner plaques in which pigmentation is the predominant feature and the induration is minimal.18
Different presentations of plaque morphea. A, A violaceous plaque characteristic of an early inflammatory lesion. B, Hyperpigmented plaques with minimal induration of the skin in the later phases. C, Indurated, ivory-colored plaque. D, Multiple, poorly defined, hyperpigmented plaques on the trunk.
Very rarely, blisters or erosions may form over the plaques of morphea, giving rise to the condition known as bullous morphea. Curiously, in the majority of cases reported, the underlying pathological change is of the lichen sclerosus et atrophicus type (Fig. 3)19 although, in some cases, the blistering may be attributed to lymphatic obstruction by the sclerotic process itself20 or to an associated autoimmune bullous disease.21
Multiple plaques in a single patient. Some are of a uniform ivory-white color, with clinical and microscopic features of morphea, and others show a mottled appearance with clinical and microscopic features of lichen sclerosus et atrophicus. Blisters and scabs are visible on some of the plaques.
The presence of microscopic features of lichen sclerosus et atrophicus in cases of bullous morphea and the rare but well-documented concomitant appearance of typical plaques of morphea and of lichen sclerosus et atrophicus in a single patient22–25 raise the question of whether lichen sclerosus et atrophicus should be considered a superficial form of plaque morphea. This is a controversial question for which there is as yet no adequate explanation. Although, as noted above, some patients develop lesions of both types, which can be difficult to distinguish clinically and microscopically, some studies have attempted to differentiate them on the basis of light-microscopic and ultrastructural features.24,26,27
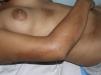
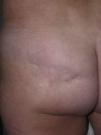
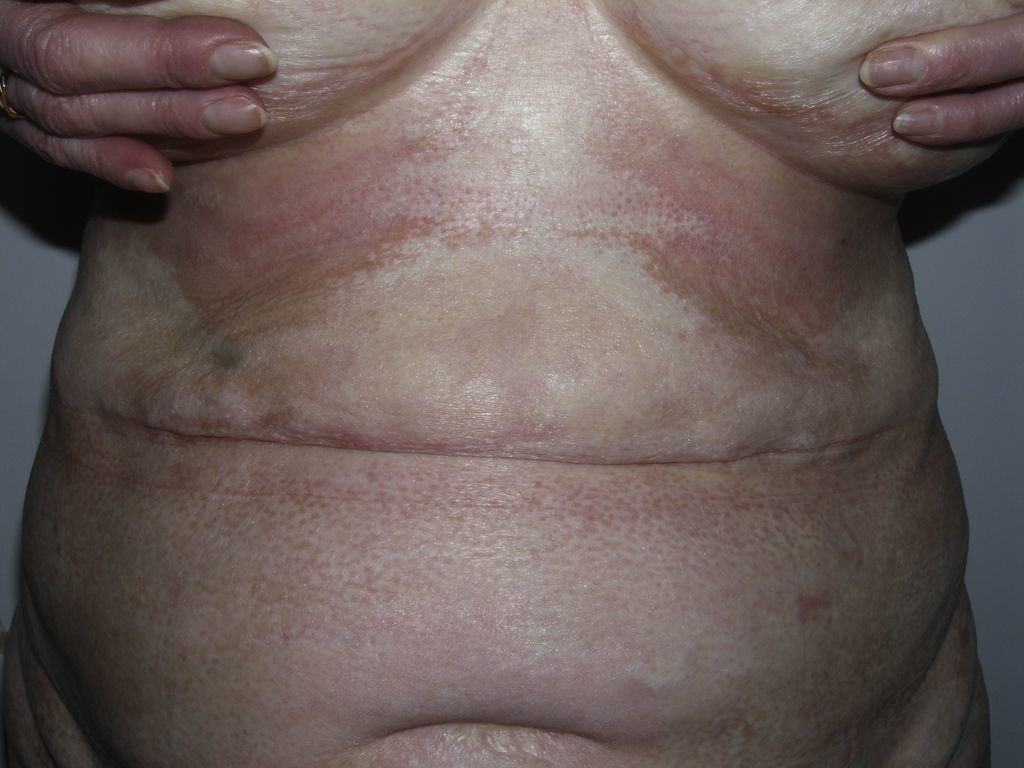
Sclerotic changes affecting a circumscribed area of skin can sometimes extend beyond the dermis to involve the adipose tissue or other underlying tissues, such as the fascia or muscle. In such cases we speak of subcutaneous morphea or solitary morphea profunda28–31 (the latter term has also been used to refer to more extensive and widespread lesions [see below]). Presentation is often as a single lesion, frequently situated on the upper part of the back, close to the midline. The overlying skin may appear normal or atrophic and may be indurated, but it is almost always depressed and adherent to the deeper tissues (Fig. 4). Blisters have occasionally been reported.32 In general, the condition is asymptomatic and is not associated with visceral alterations. The most evident microscopic findings in this form of morphea are dense sclerosis of the collagen and marked subcutaneous inflammation with the presence of lymphocytes and plasma cells; bone formation in the deep dermis has also occasionally been observed.33,34 In some cases, solitary morphea profunda or similar lesions have been reported after vaccinations35 or after intramuscular vitamin K injection.36,37
Generalized Morphea, Deep Morphea, and Eosinophilic Fasciitis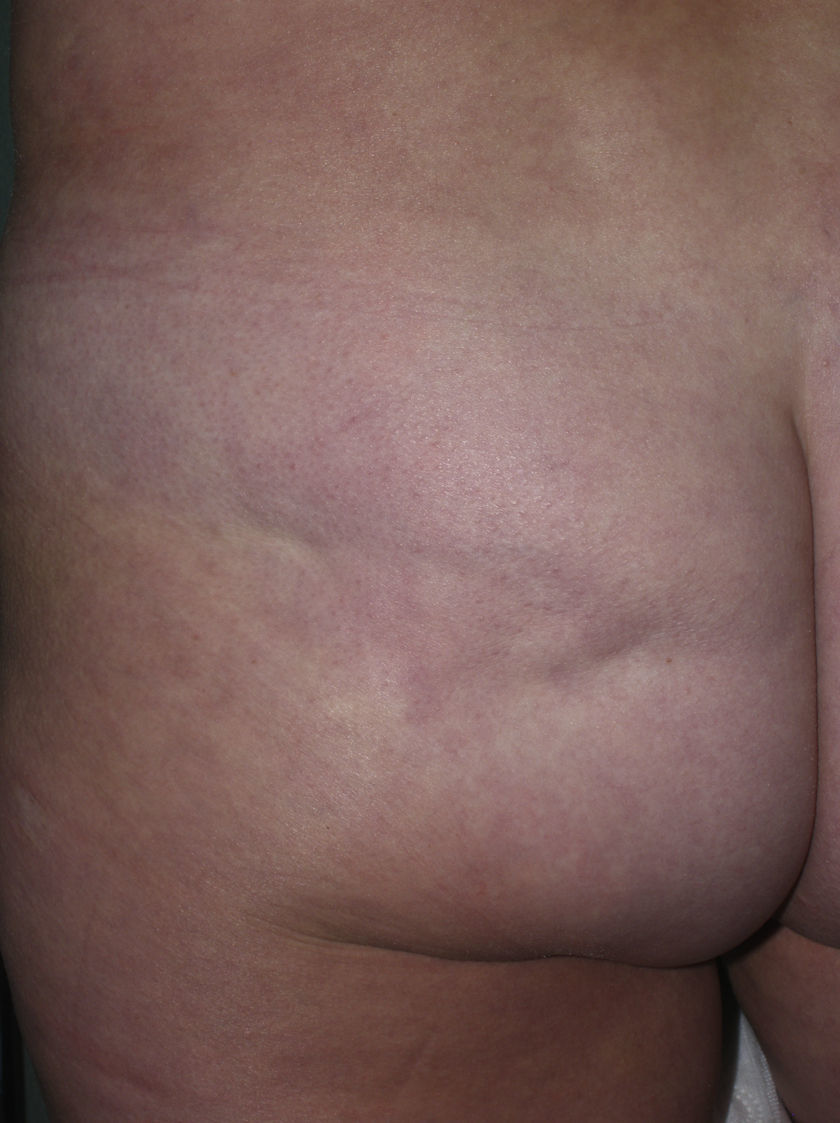
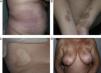
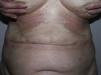
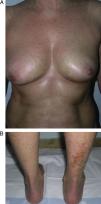
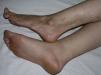
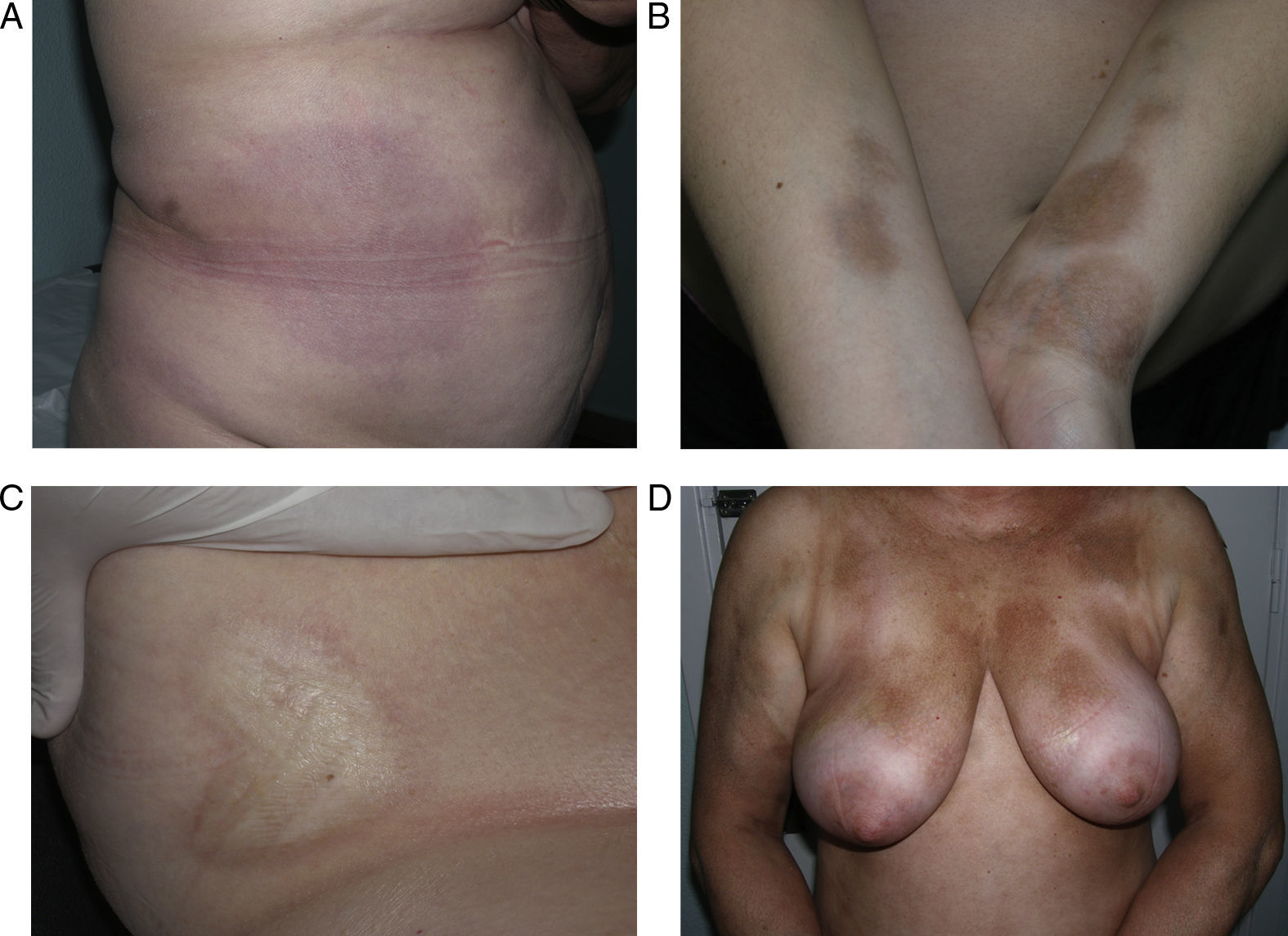
Generalized morphea is the term used to describe plaques of morphea that affect more than 2 anatomical regions. Generalized morphea has been described more often in women, and physical exercise has been proposed as a possible trigger. Manifestations include slightly inflamed, poorly defined pigmented plaques in some areas, often on the trunk, together with thickened skin adherent to the deeper planes, the fascia, and muscle in other areas, particularly on the limbs (Fig. 5). The onset of sclerosis is usually progressive and relatively rapid over a period of a few months. Acute inflammatory signs, such as edema and erythema, are typically absent. In rare cases, there may be concomitant lesions of lichen sclerosus et atrophicus and blisters may form on the surface of the plaques (Fig. 5B).20,32
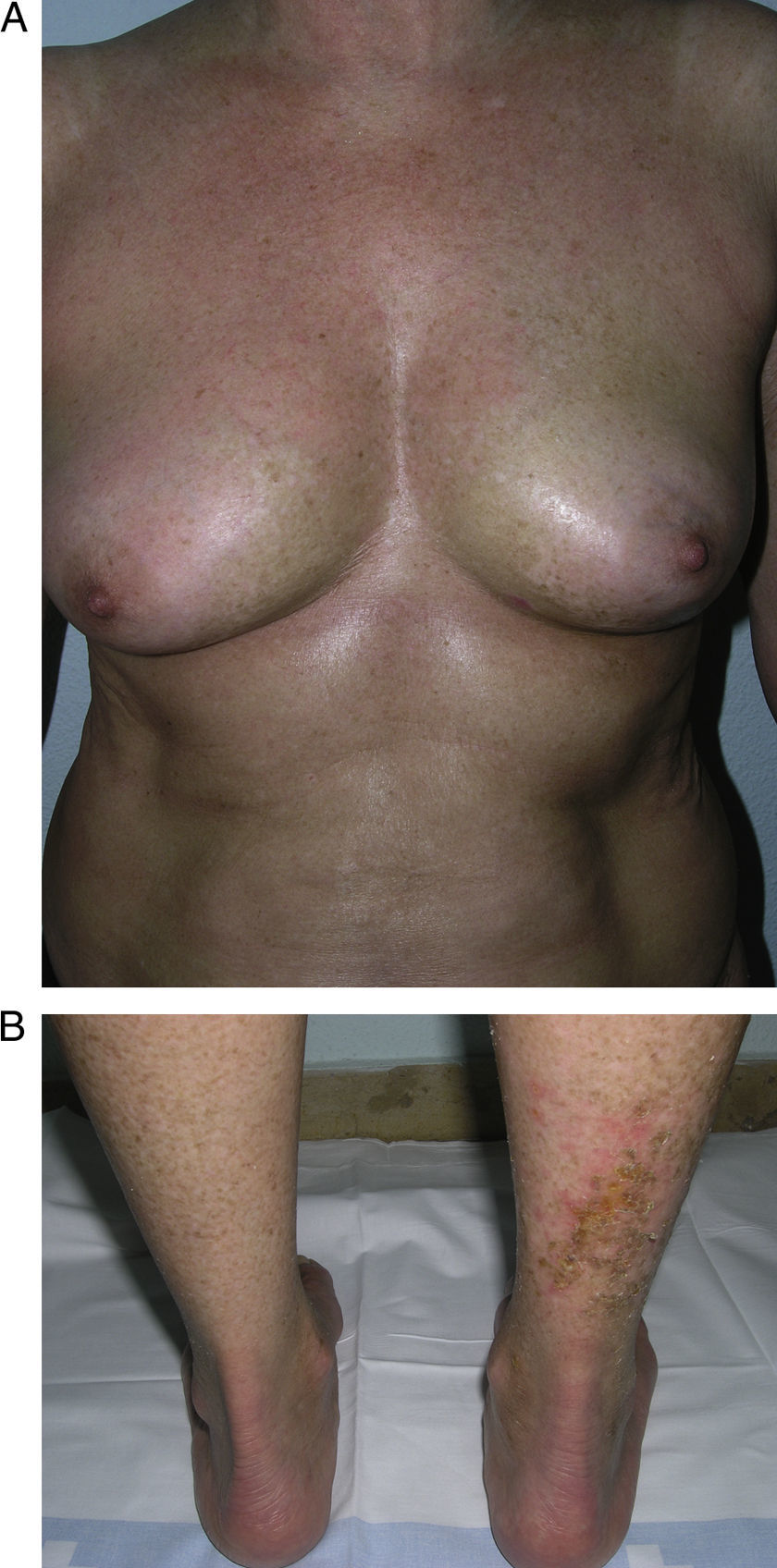
Generalized morphea. A, Multiple plaques of swollen, indurated, shiny skin. The plaques have become confluent and affect practically all the skin of the trunk and limbs. B, Erosions and scabs on the plaques on the distal areas of the lower limbs. Two biopsies taken from this patient revealed changes consistent with morphea in one and with lichen sclerosus et atrophicus in the other.
An in-depth review of the literature revealed that the terms generalized morphea and deep morphea are used indistinctly to refer to cases with this form of presentation.38–46 These terms have therefore been used in the literature to refer to the situation in which the sclerosis mainly affects the deep dermis and adipose tissue but there is also extensive involvement of the fascia and superficial muscle. The term generalized morphea alludes to the extensive fibrotic changes of this disease, whereas the term deep morphea indicates that the microscopic changes are seen mainly in the superficial muscle, fascia, adipose tissue, and deepest layers of the dermis.
Generalized morphea must be distinguished from systemic scleroderma. Although the digits can be affected in generalized morphea, ulceration, reabsorption of the phalanges, changes in the nail-fold capillaries, and Raynaud phenomenon are uncommon. Moreover, the face is not usually affected, which means that although the sclerotic changes of the skin may be very extensive in generalized morphea, the patient does not usually present the facial changes typical of systemic scleroderma, such as disappearance of the lines of facial expression, thinning of the lips, and radial lines around the mouth. However, flexion contractures of the limbs and joint and muscle manifestations are common.39,46 Very rarely, there have been reports of alterations of the lungs, esophagus, and even of the kidney and heart.4,6,46–48 Characteristic laboratory findings include peripheral eosinophilia, which may be marked, elevation of the gammaglobulins, and immunological alterations such as the presence of antinuclear and anti-single-stranded DNA antibodies, hypocomplementemia, and even antiphospholipid antibodies.39,46,49,50 Antihistone antibodies and elevation of the serum levels of type III procollagen are indicators of severe disease in localized scleroderma, and it is not uncommon to detect these alterations in the widespread, deep forms of morphea.51–54
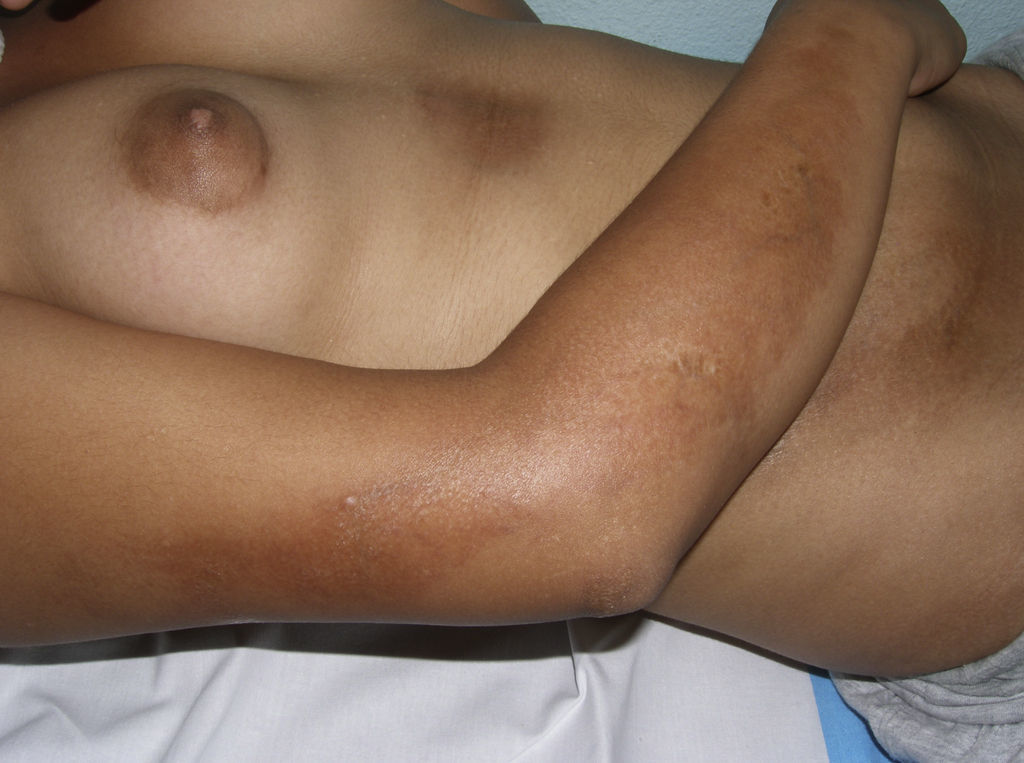
The cutaneous and systemic manifestations of generalized morphea overlap to a great extent with another disorder, eosinophilic fasciitis. The inclusion of eosinophilic fasciitis in the deep morphea group of Peterson et al.’s7 classification has been and continues to be a subject of debate in the medical literature. Without going any further, some recent publications from prestigious authors in the dermatology literature consider eosinophilic fasciitis to be a sclerodermiform syndrome.55 Nor is this disorder included in the latest classification proposed by Zulian et al.5,15 Clinically, the disease is characterized by symmetrical induration of the skin of all 4 limbs. In most patients, the condition begins with an edematous phase that produces a feeling of tightness of the skin, and diffuse areas of pale erythema appear on the limbs. This phase, which is usually of short duration and typically passes unnoticed, is followed by a phase in which the skin, now stiff and indurated, becomes irregular and dimpled, particularly over the medial surfaces of the upper limbs and of the thighs, as a result of the development of sclerotic bands that cross the subcutaneous fat perpendicularly between the deep dermis and the fascia (Fig. 6). In the final stages, the skin is completely smooth and indurated and is adherent to the deeper planes.56–59 Lesions typical of plaque morphea are observed on other areas of the body, mainly the trunk, in around 30% of patients diagnosed with eosinophilic fasciitis (Fig. 7).59–62 In general, these lesions are not synchronous with the fasciitis and can appear before or after the inflammation of the fascia. The presence of alterations in the peripheral blood, such as eosinophilia, hypergammaglobulinemia, and elevation of the erythrocyte sedimentation rate, is characteristic. However, their absence does not rule out the diagnosis as these changes are usually transient.59 Similarly, although the presence of inflammation of the fascia with an infiltrate rich in eosinophils is a characteristic microscopic finding, this feature is not essential for making the diagnosis.63 Finally, the most common extracutaneous manifestations are synovitis or tenosynovitis, arthritis, contractures, and carpal tunnel syndrome. Although visceral complications affecting the lung, esophagus, or heart have been described, they are rare. The hematological abnormalities described can be pronounced and, in addition to the eosinophilia, these patients can develop megacaryocytic aplasia,64 aplastic anemia,65–67 thrombocytopenia, hemolytic anemia,68 and myeloproliferative or lymphoproliferative disorders.69,70
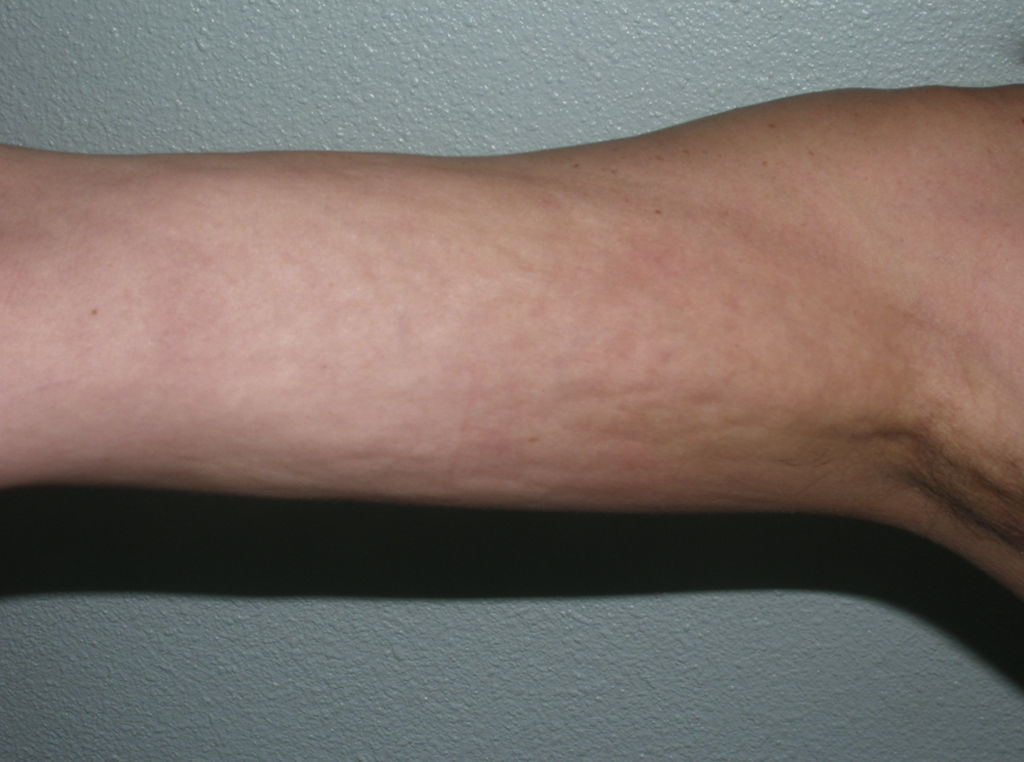
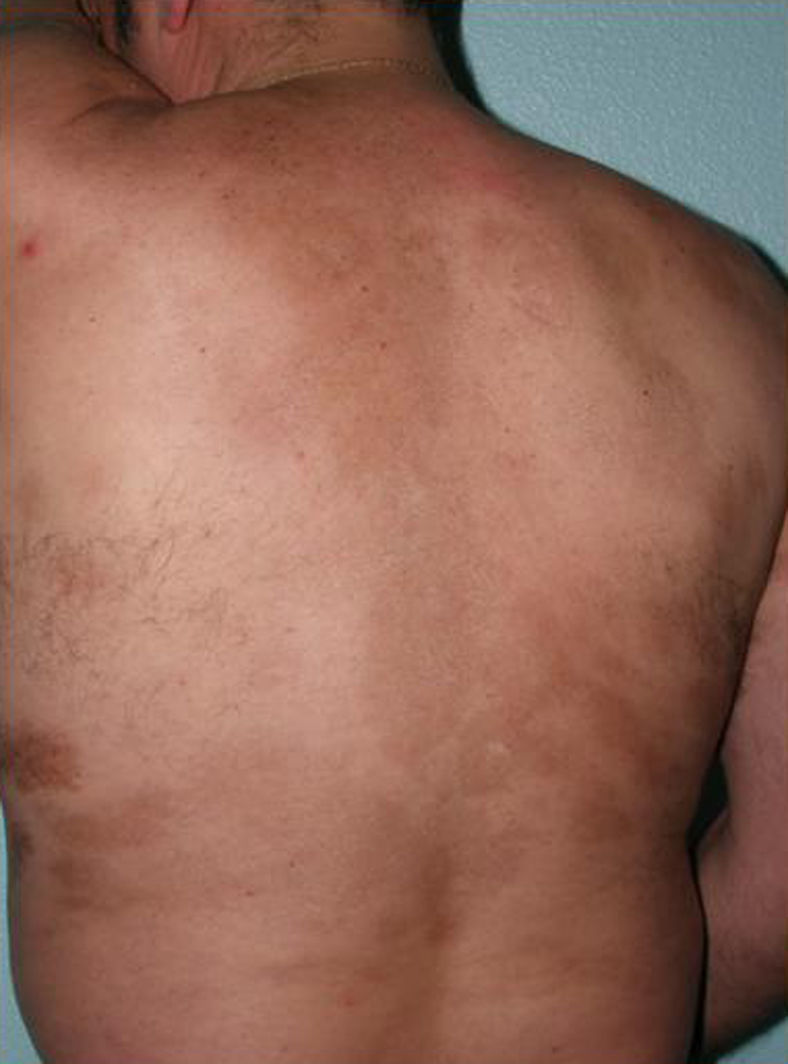
Most of the literature describing the clinical and microscopic characteristics of eosinophilic fasciitis has been published by rheumatologists, the specialists who mainly use this term. However, their descriptions include many of the cutaneous and extracutaneous features of the disease described as generalized or deep morphea in the dermatological literature. It would appear likely that all these terms (generalized morphea, deep morphea, and eosinophilic fasciitis) in actual fact refer to the same clinical entity. However, the existence of typical or “pure” cases of each disorder probably justifies the continued use of the existing terminology.
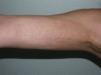
Linear MorpheaLinear morphea or linear scleroderma is often observed in childhood or youth and is probably the most common variant of morphea in these age groups, affecting between 40% and 70% of the children studied.5,6,48 Presentation is typically with a single unilateral lesion having a linear distribution, most commonly on the limbs, face, or scalp (Fig. 8). These linear lesions often follow the Blaschko lines, and genetic mosaicism has therefore been proposed as a factor that could produce this linear distribution of the sclerosis.71 The lesions are often deep and can interfere with limb growth, giving rise to deformity due to atrophy of the underlying muscle and bone, as well as joint contractures. Clinically the lesions appear as poorly defined bands of depressed skin with alterations of pigmentation.
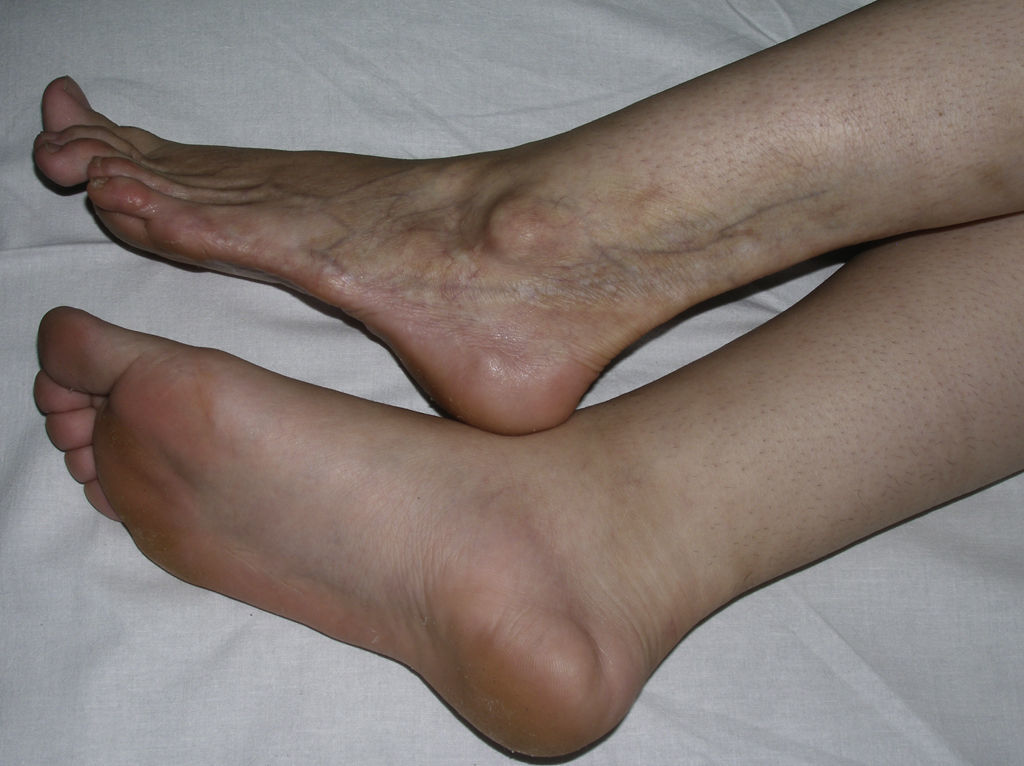
When the lesions are located on the scalp, they produce linear plaques of alopecia that are frequently atrophic and slightly depressed, and the indurated skin is smooth, shiny, and ivory-colored, though in some cases the plaques may be pigmented. The unilateral nature of these lesions, their preference for the parietal region, and their tendency to deform the bone, giving rise to depressed lesions, has led to such descriptive names as linear scleroderma en coup de sabre. Lesions can sometimes extend onto or exclusively affect the cheek, nose, or upper lip. At these sites, there is often only a mild linear pigmentation on the skin surface, but deeper involvement can produce deformities and asymmetry of facial structures and alterations of dental implantation.
When the sclerotic disorder affects a whole side of the face, it is called progressive facial hemiatrophy or Parry-Romberg syndrome. There has been a great deal of discussion about the relationship of progressive facial hemiatrophy with localized scleroderma, but the coexistence of progressive facial hemiatrophy with lesions of linear scleroderma en coup de sabre or even with plaque morphea confirms that it is a variant of linear scleroderma.72–74 The disease particularly affects adipose tissue, sometimes muscle and bone, and there is evident atrophy of the fat and muscle and facial deformities. Skin changes are almost completely absent. This atrophy favors the appearance of ocular disturbances such as endophthalmos, paralysis of the ocular muscles, ptosis, or Horner syndrome, as well as deformities of the jaw that can result in malocclusion, incorrect dental implantation, dental root atrophy, or delayed tooth eruption.74
Linear morphea, especially when the lesions are located on the head (coup de sabre and progressive facial hemiatrophy), is commonly associated with neurological complications (in almost 20% of cases),5,73,74 and ophthalmological complications (15%).75 The most significant neurological disorders are epilepsy, migraine, neuralgia and/or paresthesia of various cranial nerves, and alterations of the electroencephalogram and certain imaging studies. The predominant ophthalmological abnormality is sclerosis of the adnexal structures, followed by inflammation of the anterior segment and anterior uveitis; these latter 2 complications are often asymptomatic and unilateral.75 There is also a higher risk of other extracutaneous complications, particularly neurological disorders, when ocular lesions are present.75
Pansclerotic Morphea of ChildhoodThis is a very rare and highly aggressive and mutilating variant of deep morphea.76 It typically affects children, although onset in adult life has been reported.77 Presentation is similar to that of generalized morphea, but there is more marked and more extensive involvement not only of all the layers of the skin and subcutaneous cellular tissue but also of deeper structures such as muscle, tendons, and bone. The sclerotic plaques typically form on the extensor surfaces of the limbs and on the trunk and progress to affect all of the skin, including the face, neck, and scalp. The tips of the fingers and toes are not affected and there is no Raynaud phenomenon.76–78 The intense sclerotic changes in the skin and underlying tissues lead to pronounced joint contractures, deformity, very painful ulceration, and calcifications. Squamous cell carcinoma has occasionally be reported on long-standing pansclerotic plaques.79,80 The systemic complications and the laboratory test abnormalities observed in this disease are identical to those reported in other related disorders (eosinophilia, hypergammaglobulinemia, elevation of the erythrocyte sedimentation rate, positive antinuclear antibodies, abnormal lung function tests, esophageal disturbances, etc.).
TreatmentAs no truly effective global treatment yet exists for localized scleroderma, management must be based on the extent and the severity of the disease, and focus primarily on the risk of deformities and movement limitation.81 Treatment of these complications is difficult, and a preventive approach is therefore essential. The decision on when to start systemic therapy is hampered by the difficulty of determining clinically if the lesions are active and are going to progress or if the disease is stable and whether the damage already present will respond to treatment. It can also be difficult to qualify and evaluate any improvement in the lesions, whether in the natural course of the disease or after starting treatment. Numerous devices of differing degrees of complexity (cutometer, durometer, and ultrasound, thermographic, and computerized imaging systems, etc.) have been used to objectively measure the clinical course of morphea, but none of these approaches is universally accepted. Nor is any validated system available to score the level of activity of the disease or the damage caused. The recently published LoSCAT (Localized Scleroderma Cutaneous Assessment Tool) is a promising measurement tool as it appears to be able to differentiate between activity and damage, it is sensitive to changes, and it does not require complex equipment.82 Using the LoSCAT, the physician can quantify damage to the skin and extracutaneous tissues in a patient with localized scleroderma by evaluating a series of symptoms and clinical signs of activity (skin hardening, erythema) and damage (atrophy and alterations of pigmentation) together with the results of a number of laboratory parameters. An effective assessment tool is obviously essential if we are to conduct the multicenter studies and meta-analyses necessary for the study of rare diseases such as morphea.
The current lack of effective assessment tools explains why there are still no randomized double-blind studies to demonstrate the actual efficacy of most of the drugs proposed for the treatment of morphea. Randomized, placebo-controlled studies have shown that neither intralesional interferon83 nor oral calcitriol84 are effective systemic treatments for morphea.
The drugs most widely accepted as useful in the treatment of this disease are methotrexate and systemic glucocorticoids, almost always administered in combination. Their efficacy is supported by the results of a number of prospective85–87 and retrospective88–91 studies, but none were randomized, double-blind, or placebo controlled. Those studies included adults and children and the methotrexate dosages varied between 0.3 and 0.4mg/kg per week in children and between 15 and 25mg per week in adults. Glucocorticoids are usually administered as high-dose intravenous boluses of methylprednisolone, sometimes followed by a tapering regimen of oral prednisone. It is generally agreed that the administration of high-dose boluses of glucocorticoids achieves the desired anti-inflammatory and immunomodulator effect with a lower risk of the side effects than can develop with long-term administration of these drugs. Adults are usually administered 1g/d of methylprednisolone on 3 consecutive days per month for a maximum of 6 months, whereas the dose in children is of 30mg/kg/d of methylprednisolone (maximum dose, 500mg/d) as intravenous boluses on 3 consecutive days, repeated each week or each month.86,87 Improvement, based on the clinical impression of the physician prescribing the treatment, was observed in more than 80% of cases in the majority of those studies. When the Modified Skin Score (which has not been validated) and ultrasound measurements of the sclerosed skin were used to assess the response to treatment, only 2 of the prospective studies conducted in adults showed a significant improvement.85,86 The retrospective and uncontrolled design of those studies does not allow us to determine whether the results in the patients treated with methotrexate and glucocorticoids were similar to those of patients treated with methotrexate alone. Monotherapy with oral glucocorticoids at dosages between 0.5 and 1mg/kg/d has been shown to be effective in some studies, but there is probably a higher risk of relapse after stopping treatment.92
The various forms of UV therapy constitute another approach that must be included among the therapies available for the treatment of morphea. These include broadband UV-A therapy with or without psoralen (bath, cream, or oral), UV-A1, and narrowband UV-B. The mechanism underlying the beneficial effects of phototherapy in morphea is unknown. The majority of studies of phototherapy in morphea have used UV-A1. This form of UV radiation can induce apoptosis of Langerhans cells and T lymphocytes, reduce collagen synthesis, increase collagenase secretion, and alter the local concentrations of cytokines such as interleukin 6, transforming growth factor β, and interferon γ, which also affect collagen and glycosaminoglycan production, fibroblast growth, and the concentration of metalloproteinases in the matrix.93,94
UV-A1 is the phototherapy with the most experience and the greatest efficacy. Since Kerscher et al.95 first used UV-A1 to treat morphea in 1995, its efficacy has been evaluated in prospective studies in more than 100 patients.96–102 Improvement has been reported in 90% of patients, based on clinical examination, a skin scoring system, ultrasound, biopsy, cutometer, or a combination of these systems. In the opinion of some authors, UV-A1 therapy is particularly beneficial in the final, fibrotic phase of morphea. However, it is not very useful in patients with particularly aggressive forms of the disease characterized by involvement of the subcutaneous tissue and muscle and rapid progression.81,86,103 The main obstacles to the routine use of UV-A1 are the need for specific equipment not available in most phototherapy centers and prolonged exposure (between 30 and 60minutes, 3 times a week). In addition, the most effective treatment regimens have not yet been clearly defined. High doses of UV-A1 (130J/cm2 per session with a total dose of 3900J/cm2) are probably more effective than intermediate doses (70J/cm2 per session with a total dose of 2100J/cm2) or low doses (20J/cm2 per session with a total dose of 600J/cm2), although most patients to date have received low doses.82 Finally, it has not been demonstrated that this type of phototherapy is as effective in patients with darker skin phototypes (phototype IV or higher), as most of the studies have been performed in countries in which the majority of the population has fair skin (phototypes l to III). Wang et al.104 showed experimentally that the administration of high doses of UV-A1 produced a greater reduction of type 1 and type 3 collagen and a greater increase in the matrix metalloproteinases in patients with lighter skin phototypes. Moreover, the reduction in the production of type 1 and type 3 collagen was greater in patients treated with a single high dose of UV-A1 than in those who had received 3 sessions of high doses of UV-A1 before any assessment was made. These findings suggest that UV-A1 should be administered as pulses (a single high dose with an interval of several weeks before the following session) or else as low-dose therapy in order to prevent tanning and thus increase efficacy.
There is much less experience with other forms of phototherapy, in particular with broadband UV-A with or without psoralens, although some prospective studies have reported clinical improvement (with or without ultrasound or biopsy assessment) in around 80% of treated patients.105–107 The authors of some of those studies suggested that treatment with topical psoralen, whether by baths or cream, plus UV-A could be particularly useful in the earliest inflammatory phase of morphea.81 Experience with the use of narrowband UV-B is even more limited. The efficacy of this option has only been reported in isolated cases and we await its evaluation in controlled studies or larger case series.108
The use of other immunosuppressants for the treatment of morphea has been proposed based on their application in systemic scleroderma, though benefits have only been reported in isolated cases. This is the case with D-penicillamine, not advisable because of its poor safety profile, ciclosporin, used in 2 children with linear morphea,109 and extracorporeal photophoresis, sometimes combined with mycophenolate mofetil.110,111 Mycophenolate mofetil, an immunosuppressant that is usually well tolerated, was found to be effective in children with morphea that had not responded to treatment with a combination of glucocorticoids and metotrexate.112 That retrospective study included 10 children with various severe forms of localized scleroderma (pansclerotic morphea, generalized morphea, and linear morphea), and a clinical improvement was achieved in all patients, thus permitting the dose of glucocorticoids and methotrexate to be reduced or withdrawn. In a recent prospective study conducted in patients with diffuse systemic scleroderma, mycophenolate mofetil was shown to soften the skin.113 The good safety profile of this drug makes it a useful treatment option when the other, more traditional approaches described above proved ineffective.
Significant improvement has also been reported in cases of generalized morphea after treatment with infliximab, an anti-tumor necrosis factor agent,114 and imatinib, a tyrosine kinase inhibitor.115
Topical therapy, for which several options exist, should only be used in the most superficial and limited forms of morphea, such as plaque morphea. Topical corticosteroids, particularly high potency corticosteroids, have traditionally been recommended on morphea plaques, particularly during the early, most inflammatory phase. However, no studies have demonstrated the actual efficacy of this treatment.82 A single randomized, double-blind, placebo-controlled pilot study conducted in 10 patients with plaque morphea found tacrolimus to be effective.116 In a prospective study of 12 patients, the application of imiquimod 3 times a week was shown to be effective in reducing the erythema and induration of the plaques of morphea.117 Subsequently, imiquimod was shown to improve the plaques of morphea in 2 cases after its application 5 times a week for 16 weeks.118 Calcipotriol in combination with betamethasone has also been shown to be effective in a prospective study of 6 patients with plaque morphea.119 Finally, photodynamic therapy has been found to be totally ineffective in the treatment of morphea.120
To summarize, in Figure 9 we propose a treatment algorithm for localized scleroderma based on our own experience, that of other authors, and the most recent reviews on this topic.82
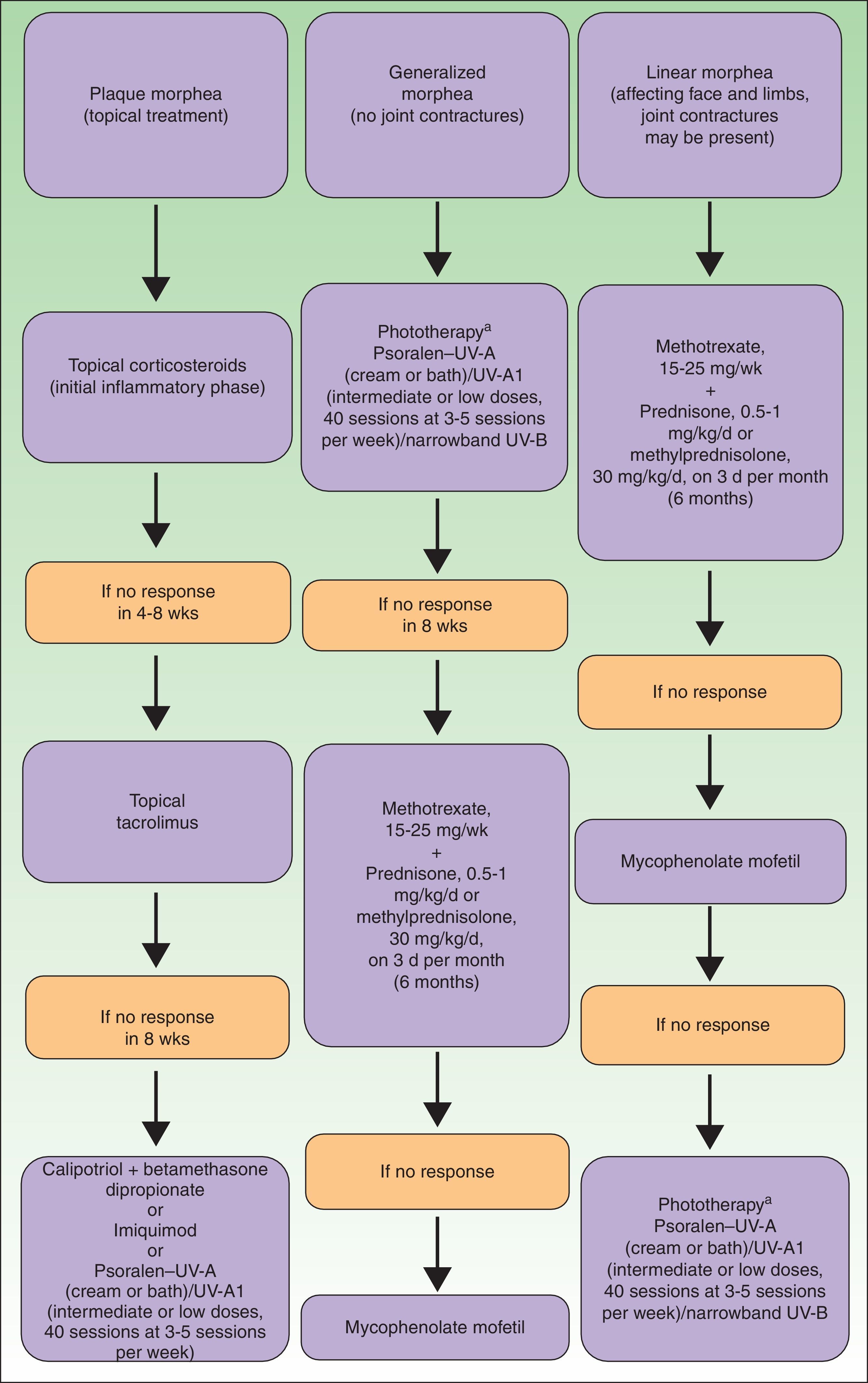
Therapeutic algorithm for localized scleroderma.81,82
aAs available.
Physiotherapy is an option worth considering in patients in whom morphea has led to joint contractures and limitations of limb movement, although once again the true usefulness of such therapy has not been demonstrated by any studies. However, it does not appear to exacerbate the disease and may therefore be prescribed.82
Whatever the therapeutic regimen used, all types of morphea tend to progress and recur, particularly when onset occurs during childhood.121 It is therefore not surprising that, throughout their lives, these patients may require various periods of treatment to cope with new outbreaks of disease activity.
Ethical DisclosuresProtection of human and animal subjectsThe author declares that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe author declares that she has followed her hospital's protocol on the publication of data concerning patients and that all patients included in the study have received sufficient information and have given their written informed consent to participate in the study.
Right to privacy and informed consentThe author obtained informed consent from the patients and/or subjects referred to in this article. This document is held by the corresponding author.
Conflicts of InterestThe author declares that she has no conflicts of interest.
Please cite this article as: Bielsa Marsol I. Actualización en la clasificación y el tratamiento de la esclerodermia localizada. Actas Dermosifiliogr.2013;104:654–66.


