





The term autosomal recessive congenital ichthyosis (ARCI) refers to a group of rare disorders of keratinization classified as nonsyndromic forms of ichthyosis. This group was traditionally divided into lamellar ichthyosis (LI) and congenital ichthyosiform erythroderma (CIE) but today it also includes harlequin ichthyosis, self-healing collodion baby, acral self-healing collodion baby, and bathing suit ichthyosis.
The combined prevalence of LI and CIE has been estimated at 1 case per 138 000 to 300 000 population. In some countries or regions, such as Norway and the coast of Galicia, the prevalence may be higher due to founder effects. ARCI is genetically highly heterogeneous and has been associated with 6 genes to date: TGM1, ALOXE3, ALOX12B, NIPAL4, CYP4F22, and ABCA12. In this article, we review the current knowledge on ARCI, with a focus on clinical, histological, ultrastructural, genetic, molecular, and treatment-related aspects.
Las ictiosis congénitas autosómicas recesivas (ICAR) son trastornos infrecuentes de la queratinización que se engloban en las formas no sindrómicas de ictiosis. Clásicamente se distinguían en este grupo la ictiosis laminar (IL) y la eritrodermia ictiosiforme congénita (EIC). Actualmente se incluyen también la ictiosis arlequín, el bebé colodión autorresolutivo, el bebé colodión autorresolutivo acral y la ictiosis en traje de baño.
Se ha estimado una prevalencia conjunta para IL y EIC de 1:138.000-1:300.000. En algunos países o regiones, como Noruega y la costa gallega, la prevalencia podría ser mayor debido a la existencia de efectos fundadores. Desde el punto de vista genético son muy heterogéneas. Seis genes se han asociado a estas entidades: TGM1, ALOXE3, ALOX12B, NIPAL4, CYP4F22 y ABCA12. En este trabajo se pretenden revisar los conocimientos actuales en el campo de las ICAR, incluyendo aspectos clínicos, histológicos, ultraestructurales, genético-moleculares y de tratamiento.
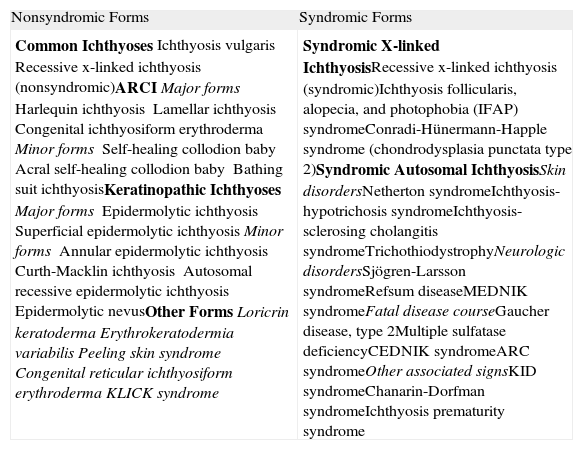
The latest consensus classification of ichthyosis differentiates between 2 main forms: the nonsyndromic forms, which present with skin manifestations only, and the syndromic forms, which present with manifestations in other organs as well (Table 1).1 Among the nonsyndromic forms, 4 groups are identified: common ichthyoses, autosomal recessive congenital ichthyoses (ARCIs), keratinopathic ichthyoses, and other less common ichthyoses.Traditionally, the group of ARCIs was divided into 2 disorders, lamellar ichthyosis (LI) and congenital ichthyosiform erythroderma (CIE). In the new classification, harlequin ichthyosis (HI) was added to this group1 because inactivating mutations in the ABCA12 gene have been identified as responsible for this disorder,2,3 while nonsense mutations in the same gene may give rise to the LI4 or CIE5,6 phenotype. Other less common variants included in the group of ARCIs are self-healing collodion baby (SHCB), acral SHCB, and bathing suit ichthyosis.7–9
Consensus Classification Based on the Clinical Features of Ichthyosis1.
| Nonsyndromic Forms | Syndromic Forms |
| Common IchthyosesIchthyosis vulgarisRecessive x-linked ichthyosis (nonsyndromic)ARCIMajor formsHarlequin ichthyosisLamellar ichthyosisCongenital ichthyosiform erythrodermaMinor formsSelf-healing collodion babyAcral self-healing collodion babyBathing suit ichthyosisKeratinopathic IchthyosesMajor formsEpidermolytic ichthyosisSuperficial epidermolytic ichthyosisMinor formsAnnular epidermolytic ichthyosisCurth-Macklin ichthyosisAutosomal recessive epidermolytic ichthyosisEpidermolytic nevusOther FormsLoricrin keratodermaErythrokeratodermia variabilisPeeling skin syndromeCongenital reticular ichthyosiform erythrodermaKLICK syndrome | Syndromic X-linked IchthyosisRecessive x-linked ichthyosis (syndromic)Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndromeConradi-Hünermann-Happle syndrome (chondrodysplasia punctata type 2)Syndromic Autosomal IchthyosisSkin disordersNetherton syndromeIchthyosis-hypotrichosis syndromeIchthyosis-sclerosing cholangitis syndromeTrichothiodystrophyNeurologic disordersSjögren-Larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, keratoderma.
Only limited data are available on the epidemiology of ARCIs. In the United States, a prevalence at birth of 1 per 100 000 population for LI and of 1 per 200 000 population for CIE has been estimated. Other studies have reported a combined prevalence for LI and CIE of 1 per 200 000 to 300 000 population.10,11 In some countries such as Norway, the estimated prevalence is greater (1 per 91 000) due to founder mutations.12 The finding of 1 or several recurrent mutations in a population may be because the mutation occurred at a given point in history and was then passed from generation to generation (founder mutation) or because the region of the genome where the mutation is found has a DNA sequence susceptible to mutation (mutation hotspot). In Spain, the estimated prevalence of ARCI is 1 per 138 000 in the general population and 1 per 61 700 among children under 10 years of age.13 In certain regions of Spain, the prevalence might be even higher. On the Galician coast, for example, a prevalence of 1 per 33 000 was reported, due also to a founder effect.14
Lamellar Ichthyosis and Congenital Ichthyosiform ErythrodermaClinical CharacteristicsAlthough it was originally thought that LI and CIE were different entities, there have been reports of patients with intermediate clinical manifestations and both conditions can be caused by mutations in the same gene.15,16 In addition, patients with the same mutation, even within the same family, can develop different phenotypes.12,15
Most patients are born enveloped in a collodion membrane that progressively disappears during the first weeks of life and is replaced by the definitive phenotype (Fig. 1A). Hypohidrosis, severe heat intolerance, and nail dystrophy are frequently observed in both LI and CIE.17–19 Patients with LI usually have more severe clinical manifestations than those with CIE. They have large platelike scales, often of a dark color, covering the whole body surface area. Erythroderma is either absent or minimal. Such patients usually have ectropion and, at times, eclabium, hypoplasia of joint and nasal cartilage, scarring alopecia, especially at the edge of the scalp, and palmoplantar keratoderma (Fig. 1B and C). CIE is characterized by the presence of erythroderma and fine whitish scaling (Fig. 2). Some patients have marked erythema and generalized scaling. The scales can be large and dark colored, particularly on the extensor surfaces of the legs. In less severe cases, erythema is mild and the scaling is fine.


Histopathologic changes do not provide a diagnosis. In LI, massive orthokeratotic hyperkeratosis is observed, usually with twice the extension as in CIE. The epidermis is acanthotic and occasionally takes on a psoriasis-like appearance. The cell proliferation rate is normal or slightly elevated.17–19 Patients with CIE have less marked hyperkeratosis, with focal or extensive parakeratosis, a normal or thickened granular layer, and more pronounced acanthosis. The epidermal turnover is increased.17–19
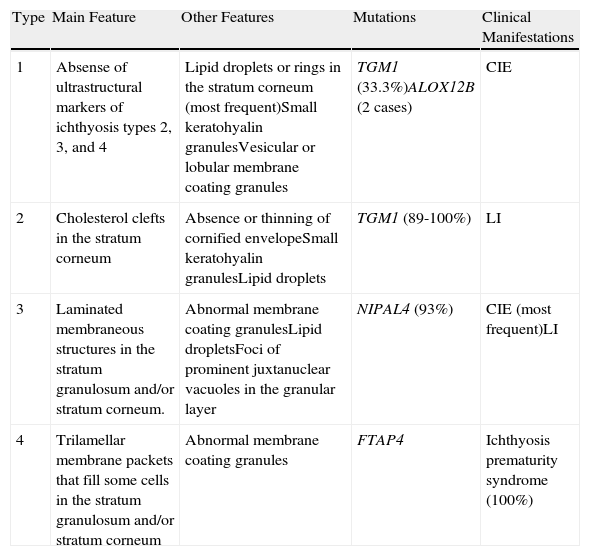
UltrastructureAlthough a close correlation between molecular, clinical, and ultrastructural findings has so far not been found, electron microscopy may nevertheless be useful for ruling out other forms of ichthyosis and for guiding genetic analyses in some cases. Four types of congenital ichthyosis have been described (Table 2).
Ultrastructural Classification of Congenital Ichthyoses.
| Type | Main Feature | Other Features | Mutations | Clinical Manifestations |
| 1 | Absense of ultrastructural markers of ichthyosis types 2, 3, and 4 | Lipid droplets or rings in the stratum corneum (most frequent)Small keratohyalin granulesVesicular or lobular membrane coating granules | TGM1 (33.3%)ALOX12B (2 cases) | CIE |
| 2 | Cholesterol clefts in the stratum corneum | Absence or thinning of cornified envelopeSmall keratohyalin granulesLipid droplets | TGM1 (89-100%) | LI |
| 3 | Laminated membraneous structures in the stratum granulosum and/or stratum corneum. | Abnormal membrane coating granulesLipid dropletsFoci of prominent juxtanuclear vacuoles in the granular layer | NIPAL4 (93%) | CIE (most frequent)LI |
| 4 | Trilamellar membrane packets that fill some cells in the stratum granulosum and/or stratum corneum | Abnormal membrane coating granules | FTAP4 | Ichthyosis prematurity syndrome (100%) |
Abbreviations: CIE, congenital ichthyosiform erythroderma; LI, lamellar ichthyosis.
Congenital ichthyosis type 1 is characterized by the absence of ultrastructural markers for ichthyosis types 2, 3, and 4. Therefore, diagnosis is usually only made when the other types have been excluded. The most frequent finding is the presence of lipid droplets or rings in the stratum corneum (Fig. 3A).20 These lipid droplets are not a constant feature or specific to this particular type as they are not present in all cases,20 and they may be present in other types of ichthyosis.21,22 Clinically, most patients present with manifestations of CIE.12,20 One-third of patients have mutations in the TGM1 gene.16 This ultrastructural type has also been identified in association with mutations in the ALOX12B gene.23,24
Congenital Ichthyosis Type 2 in corneocytes.")
Congenital ichthyosis type 2 is characterized by cholesterol clefts in the stratum corneum (Fig. 3B).21 Such clefts are a constant finding in this type of ichthyosis, and can be detected in different biopsies in the same patient; treatment with oral retinoids has no impact on these clefts.12,25 Electron-dense aggregates have also been observed on corneocytes in some patients with deficient TGase 1 activity.26–28 Clinically, most patients present with severe manifestations of CIE.12 This ultrastructural type is strongly associated with mutations in the TGM1 gene.12,16
Congenital Ichthyosis Type 3Congenital ichthyosis type 3 is characterized by lamellar membranous structures in the stratum granulosum and/or stratum corneum. These structures are arranged in strips around an empty space close to the nucleus.22,29–31 The clinical manifestations in this type are different to the others; onset of ichthyosis is variable, desquamation and erythema may be patchy or generalized, and the flexures in particular are affected. Mutations in the NIPAL4 gene are responsible for 93% of ichthyoses type 3.32
Congenital Ichthyosis Type 4Characteristically, in congenital ichthyosis type 4, some cells in the stratum granulosum and stratum corneum are filled with trilamellar membrane packages.33 These findings are pathognomic for ichthyosis prematurity syndrome, a condition currently considered as a syndromic form of ichthyosis.34,35
Molecular StudiesIn genetic terms, the ARCIs are very heterogeneous. The TGM1 gene is associated with most cases, but mutations in 5 other genes (ALOX12B, ALOXE3, NIPAL4, CYP4F22, and ABCA12) have been reported. Fischer et al.36 studied 520 families with ARCI and identified mutations in at least 1 of these genes in 78% of cases (TGM1 in 32%, NIPAL4 in 16%, ALOX12B in 12%, CYP4F22 in 8%, ALOXE3 in 5%, and ABCA12 in 5%). In another study of 250 patients with ARCI of different origins, 38% had TGM1 mutations, 6.8% had ALOXE3 mutations, and 6.8% had ALOX12B mutations.37 In Galicia, we identified mutations in the TGM1, ALOX12B, ALOXE3, NIPAL4, and CYP4F22 genes in 75% of the families studied, but the distribution of mutations was different.14 The TGM1 gene was mutated in 68.7% of the cases while the ALOXE3 gene was mutated in just 1 patient. We did not detect mutations in any of the other 3 genes studied.
TGM1The TGM1 gene is located on chromosome 14q11.2 and has 15 exons (GenBank NM-000359.2). It encodes the TGase 1 enzyme, which is one of the 3 TGase enzymes found in the epidermis.38 This enzyme participates in the formation of the cornified envelope by catalyzing calcium-dependent cross-linking of several proteins such as involucrin, loricrin, and proline-rich proteins.39,40 It also catalyzes binding of ¿-hydroxyceramides in the outer layer of the cornified envelope with proteins in the inner layer.41,42 In patients with TGM1 mutations, the cornified envelope is missing and TGase 1 activity is reduced or nonexistent.43–47
Since 1995, when this gene was identified as responsible for some cases of ARCI,48–50 more than 110 mutations have been reported in patients of different origins. Mutations in TGM1 are the most common cause of ARCI.36,37 This mutation has been found in 55% of cases in the United States and in 84% of cases in Norway.12,51 The most frequent mutation is c.877-2A>G, which has been found in 34% of the mutated alleles reported to date.52 The high frequency of this mutation in countries such as the United States and Norway is due to a founder effect.12,53 The second most frequent mutation is p.Arg142His. This and similar mutations have been reported in countries such as Egypt, Germany, Finland, and the United States,15,49–51,54–56 and it would seem that these are hotspot mutations.57 The p.Arg307Trp mutation is frequent in the Japanese population.5 In Galicia, the p.Arg760X, c.1223_1227delACACA and c.984+1G>A mutations in TGM1 were identified in 81.82% of the families with mutations in this gene, suggesting a founder effect.14 Confirmation of this hypothesis was obtained by haplotype study (work as yet unpublished).
TGM1 mutations are responsible for most cases of LI15,27,44,46,56,58–63 and for a small percentage of cases of CIE.43,47,64,65 Such mutations can also give rise to other forms of ARCI such as SHCB, acral SHCB, and bathing suit ichthyosis.
Many studies have attempted to demonstrate genotype-phenotype associations between mutations in TGM1 and ultrastructural or clinical findings, but no significant correlation has been observed to date.15,16,53 In general, patients with mutations in the TGM1 gene are more severely affected than those without such mutations. In a study of 83 patients with ARCI in Sweden and Estonia, the presence of ectropion and collodion baby was associated with TGM1 mutations, while a higher rate of erythema was observed in patients without mutations in this gene.66 Another study showed that the type of scaling is the main difference between carriers and noncarriers of TGM1 mutations, on finding that all patients with mutations in this gene had lamellar scaling whereas 80% of those without TGM1 mutations had fine scaling.14 In addition, it has been seen that truncating mutations are more frequently associated with hypohidrosis and sweating disorders than missense mutations.51 In the north American population, a model based on the presence of certain clinical characteristics predicts that patients who are born as collodion babies and have ocular disorders and/or alopecia are 4 times more likely to have TGM1 mutations.51
ALOXE3 and ALOX12BThe ALOXE3 and ALOX12B genes are located on chromosome 17p13.1.67 They have a similar structure with 15 exons that encode the epidermal LOXs eLOX-3 and 12R-LOX.68,69 The fact that they are predominantly expressed in the suprabasal layers of the epidermis supports their role in advanced phases of epidermal differentiation, with participation in the processing of lamellar bodies.24,70 These enzymes act on adjacent steps in the hepoxilin pathway (Fig. 4). 12R-LOX transforms arachidonic acid to 12R-hydroxyeicosatetraenoic acid while eLOX-3 converts this product into an epoxyalcohol isomer69,71 of the hepoxilin A3 family.72 The hepoxilin product is unstable and is hydrolyzed in cells to a specific trihydroxy derivative (trioxilin). Although the exact role of the products of the hepoxilin pathway is not known, it has been speculated that they may participate in the formation of intercellular lipids of the stratum corneum or act as signals for inducing keratinocyte differentiation.

The ALOX12B and ALOXE3 genes were first identified in 2002.73,74 Since then, more than 30 mutations in the ALOX12B gene23,24,37,75–77 and approximately 10 in the ALOXE3 gene37,74,75 have been reported. These mutations are responsible for 14% to 17% of ARCIs36,37 and 72.2% of SHCBs.23,78,79 The causative relationship between these mutations and phenotype was confirmed by demonstrating that the catalytic activity of the epidermal LOX was totally abolished in patients with these mutations75,80 and by using animal models that reproduced the ichthyosiform phenotype seen in humans.81–83 Both genes are responsible for a similar percentage of ARCI cases. However, the range of different mutations in the ALOXE3 gene is limited, due to the predominance of 2 mutations, p.Arg234X and p.Pro630Leu, which seem to correspond to hotspots.37,74,75
The patients with mutations in the ALOXE3 and ALOX12B genes usually show a CIE phenotype.74,75,77 The severity of scaling is mild or moderate, and the scales have a whitish or light brown color. Erythema may also be present. As many as 76% of the patients are born as collodion babies and 88% have sweating disorders.37 Patients with mutations in the ALOX12B gene show more limited, whitish desquamation compared with carriers of mutations in the ALOXE3 gene. In these cases, the scales are brownish and adherent. The presence of erythema, palmoplantar hyperkeratosis, and accentuation of the palmoplantar folds are also associated with ALOX12B mutations.37
Ichthyin/NIPAL4The NIPAL4 gene, also known as the ichthyin gene, is located on chromosome 5q33. It has 6 exons that encode a protein with several transmembrane domains of unknown function.84 It has been hypothesized that the protein product participates in the same metabolic pathway as LOX and may act as a receptor for trioxilins A3 and B3 or for other metabolites of the hepoxilin metabolic pathway.84 It would thus be implicated in the formation of lamellar bodies or in their transport towards the extracellular space.32 In support of this are 2 observations. First, in 93% of the cases, mutations in this gene are associated with an ultrastructural pattern of congenital ichthyosis type 3, characterized by abnormalities in the lamellar bodies and the presence of elongated perinuclear membranes in the stratum granulosum.32 Second, NIPAL4 is expressed essentially in the stratum granulosum of the epidermis, where the lamellar bodies are present.85
Since the discovery of the NIPAL4 gene in 2004,84 only 9 mutations have been reported in patients from Mediterranean countries (Algeria, Turkey, and Syria),84 Scandinavian countries,32 Pakistan,85 the Faroe Islands,32 and South America.84
The clinical spectrum of patients with mutations in this gene is broad, even among members of the same family. Between 3.7%32 and 60%84 are born as collodion babies. When the collodion membrane disappears, most patients develop the manifestations of CIE, with fine whitish scales on an erythematous base on the face and trunk and larger, brownish scales on the neck, buttocks, and legs.84 Marked xerosis, generalized brownish reticular hyperkeratotic plaques that appear accentuated in the skin folds, and facial dyschromia may be present.32,85 In addition, palmoplantar keratoderma is a frequent finding along with occasional finger contractures and curved finger nails. Some studies have reported findings more typical of LI.32,85 The presence of signs and symptoms of atopic dermatitis has been reported in some patients, although mutations in the FLG gene were not detected in any of these cases.85
CYP4F22The FLJ39501 or CYP4F22 gene is located on chromosome 19p13.12.86 It has 12 exons87 and encodes a P450 cytochrome, family 4, subfamily F, polypeptide 2, homolog of leukotriene B4- ω-hydroxylase (CYP4F2). The reaction catalyzed by the product of FLJ39501 in the skin and the substrates of that reaction may be deduced by analogy with its known homologs CYP4F2 and CYP4F3.88 It has been hypothesized that CYP4F2 and CYP4F3 participate in the hepoxilin pathway by catalyzing the conversion of trioxilin A3 to 20-hydroxy-(R)trioxilin A387 and that the end product of this pathway, 20-carboxy-trioxilin A3, may have a key biological regulatory effect in the skin.89
To date, only 8 mutations of this gene have been reported in 12 consanguineous families from Mediterranean countries87 and in 1 family of Israeli origin.62
In the families reported by Lefèvre et al.,87 most patients had a CIE phenotype at birth and this subsequently progressed to LI. Patients were usually born with marked erythroderma, although without any collodion membrane. As they got older, they developed generalized whitish-grey scaling, which was more marked in the periumbilical region, on the buttocks, and on the lower part of the body. Hyperlinearity of the palms and soles and desquamation on the scalp, at times of pityriasiform type, were frequent.87 In another family, the 3 members affected were born as collidion babies and developed intense erythroderma, generalized desquamation, and palmoplantar keratoderma.62
ABCA12In 2003, the ABCA12 gene was reported to be responsible for some cases of LI and was mapped to chromosome 2q34.4 It was subsequently confirmed that mutations in this gene were also responsible for HI.2,3ABCA12 encodes 53 exons, and belongs to a family of ABC transporters, which bind adenosine triphosphate while also facilitating the transport of several molecules across the cell membrane.90 The members of the ABCA subfamily are all implicated in lipid transport.91 Deficient ABCA12 function causes lipid transport disorders in lamellar bodies and so lead to a decrease in intercellular lipid levels in the stratum corneum.3Ultrastructural studies have shown that ABCA12 is located in lamellar bodies associated with glycosylceramides.91ABCA12 mutations have been associated with disorders in the distribution and transport of glycosylceramides and with decreased levels of hydroxyceramides, one of the main components in the lipid barrier in the intercellular spaces.3,6,92,93 The massive hyperkeratosis that occurs in these patients could be a compensatory response to a deficient lipid barrier.94 It might also be due to the lack of desquamation of the corneocytes,93 which could be caused by defects in the transport of certain proteases, such as callicrein 5 and cathepsin D, resulting from disorders in the lamellar bodies.95 Murine models and in vitro studies suggest that ABCA12 mutations also have an effect on epidermal differentiation.95–97
To date, more than 50 mutations have been reported in the ABCA12 gene in patients with ARCI from Africa, Europe, Pakistan, and Japan. The most frequent mutations are p.Val244SerfsTer28,2,98,99 identified in Pakistani and Indian populations, and p.Asn1380Ser,4 identified in African families. In both case, these may be founding mutations.
The extent of the ABCA12 mutations is related to phenotype, with mutations associated with complete loss of function leading to the HI phenotype.2,3,98–102 By contrast, in LI and CIE, most mutations are missense, and have a less severe effect on protein function.4–6,103 The mutations underlying the LI phenotype seem to be concentrated in the first adenosine triphosphate binding cassette region.4 Clinically, patients with CIE and mutations in the ABCA12 gene have medium-size scales that are somewhat larger than those usually observed in patients with this phenotype.
Harlequin IchthyosisHI or harlequin fetus is a severe and usually fatal form of ichthyosis. The children are usually premature with extensive shiny hyperkeratotic plaques, separated by deep fissures, that cover the entire integument and form geometric patterns reminiscent of clothing worn by harlequins, thereby giving the condition its name. Skin tightness leads to marked eversion of the eyelids and lips, rudimentary development of joint and nasal cartilage and, occasionally, microcephaly. The children rarely have eyelashes or eyebrows, although the hair on the scalp may be conserved. The hands and feet are swollen and edematous, and often covered by a glove-like layer. They may have finger contractures.
For such patients, the risk of dying during the neonatal period is very high.104 Pulmonary ventilation is compromised; transepidermal water loss leads to dehydration, hydroelectric imbalance, and thermal instability; and the risk of infections is increased. Facial tightness and eclabium hinder sucking and therefore feeding, with the corresponding worsening of dehydration. Neonates with this condition rarely lived longer a few weeks. In recent years, however, the chances of long-term survival have increased notably, essentially due to administration of systemic retinoids and progress in intensive neonatal care.105 In a recent study, 83% of the patients treated with oral retinoids survived compared to 24% of untreated patients. Most of the deaths occurred in the first 3 days of life, but treatment was not started until after this in many of the survivors.104 This would suggests that many of these early deaths would have occurred regardless of retinoid treatment.
The children who survive the neonatal period generally develop severe CIE.106 The nature and location of mutations in the ABCA12 gene and the extent of transporter function loss may determine prognosis.3,92,107 Patients who conserve a certain degree of protein activity, albeit minimal, may have a better chance of surviving. Carriers of homozygous mutations have a higher mortality rate.104
The main histologic characteristic of HI is the presence of an extremely thick and compact orthokeratotic stratum corneum. The hair follicles and sweat ducts have prominent hyperkeratotic plugs107,108 and have abnormal or absent lamellar bodies, lipid inclusions, or remnants of organelles or nuclei in the corneocytes, and absence of intercellular lipids in the ultrastructural study.108,109 The hair follicles show a marked concentration of keratotic material, which is a diagnostic feature of HI used for prenatal diagnosis.
To date, the rate of detection of mutations in the ABCA12 gene in patients with HI is close to 100%, and so this would appear to be a genetically homogeneous condition.
Collodion Baby and Self-healing Collodion BabyCollodion babies are usually born prematurely and perinatal morbidity and mortality are increased. At birth, the neonate is covered by a shiny taught transparent membrane reminiscent of cellophane wrapping (Fig. 5). The babies have ectropion, eclabium, and hypoplasia of the nasal and joint cartilage. Sucking and pulmonary ventilation may be hindered110 and transepidermal loss of water and the risk of infections are increased.110,111

Collodion baby is the usual presentation for HI and CIE. Autosomal dominant LI,112,113 Sjögren-Larsson syndrome,110 trichothyodystrophy,114 juvenile Gaucher disease,110 neutral lipid storage disease, Conradi-Hünermann-Happle syndrome, Hays-Wells syndrome, and ectodermal dysplasia115 may also occasionally present as collodion baby. The membrane disappears spontaneously in 10% to 24% of neonates, to give way to completely normal skin.110,116 In the past, these cases were described as LI of the newborn,117 but they are not referred to as SHCB.118 Some authors have suggested the term self-improving collodion ichthyosis because many of these patients, when reexamined later in childhood or as adults, have a variable degree of anhidrosis and heat intolerance and mild signs of ichthyosis, such as xerosis and fine desquamation, particularly in the axillae and neck.78
Neither optical microscopy nor ultrastructural investigations of collodion baby are specific. It is therefore preferable to delay the skin biopsy until the definitive phenotype has developed.
Mutations in the TGM1,7,119ALOXE3,78 and ALOX12B23,78,79 genes have been identified in patients with SHCB. ALOX12B mutations are the most common. In a series of 15 Scandinavian patients with SHCB, 67% had mutations in the ALOX12B gene, 25% in the ALOXE3 gene, and 8.3% in the TGM1 gene.78 Mutations were not found in some patients, and so other genes are also likely to be implicated. There has been speculation that these mutations reduce enzymatic activity in the uterus but not after birth.7 In the uterus, where the hydrostatic pressure is high, chelation by water converts the mutated enzyme into an inactive conformation. After birth, when the pressure decreases, the enzyme returns to its active form and its activity increases sufficiently to maintain a normal or minimally affected phenotype.7
Acral Self-healing Collodion BabyAlthough collodion baby affects the whole body, cases confined to the acral regions have been reported. In 1952, Finlay et al.120 reported a case of collodion membrane that affected only the hands and feet and that followed a self-healing course. Recently, a new case of acral SHCB has been reported in association with mutations of the TGM1 gene.8 It is not known why these lesions are restricted to acral regions, although factors associated with site-dependent regulation of enzyme activity may be in operation.8
Bathing Suit IchthyosisBathing suit ichthyosis was first reported as an independent ARCI variant in 2005 although cases of ichthyosis with a peculiar distribution had been reported previously.121–123 It has been detected mainly in patients of South African origin,9 although it has also been reported in individuals from Europe and Mediterranean countries.124 At birth, patients have a generalized collodion membrane which then sheds to leave the characteristic distribution of scaling. The trunk, proximal region of the arms, including the axillae, the neck, and the scalp are generally affected, while the central part of the face, the limbs, and the adrenal region are usually spared.9 The scales are large, lamellar, and dark in color. Finer desquamation may occur in the popliteal and antecubital fossae.124,125 The palms of the hands and soles of the feet have mild diffuse hyperkeratosis whereas the backs of the hands and feet show no involvement.
Histopathologic study of affected skin shows marked hyperkeratosis without parakeratosis, normal granular layers, mild or moderate acanthosis, and a mild lymphocytic infiltrate in the upper dermis.9 Electron microscopy observations are consistent with congenital ichthyosis type 2 in most cases. Uninvolved skin does not show any abnormal findings.124,125 In healthy skin, TGase 1 activity is slightly reduced and usually localized in pericellular areas. In involved skin, enzymatic activity is residual and abnormally located in the cytoplasm.124
Mutations have been detected in the TGM1 gene in all patients with bathing suit ichthyosis studied to date.119,124–126 The most common mutation is p.Arg315Leu, which has been identified in most South African patients and could be a founding mutation. Oji et al.124 suggested that skin temperature might play a role in the development of these manifestations. Using digital thermography, the authors showed a strong correlation between body temperature and desquamation, with the hottest areas of the body being the ones most affected. Aufenvenne et al.127 showed a decrease in optimum temperature for TGase 1 activity in patients with bathing suit ichthyosis. This decrease was not observed in healthy controls or in patients with generalized LI. This decrease in temperature would explain the phenotype of these patients. The optimum temperature is 37°C for the normal enzyme but 31°C for the mutated enzyme.
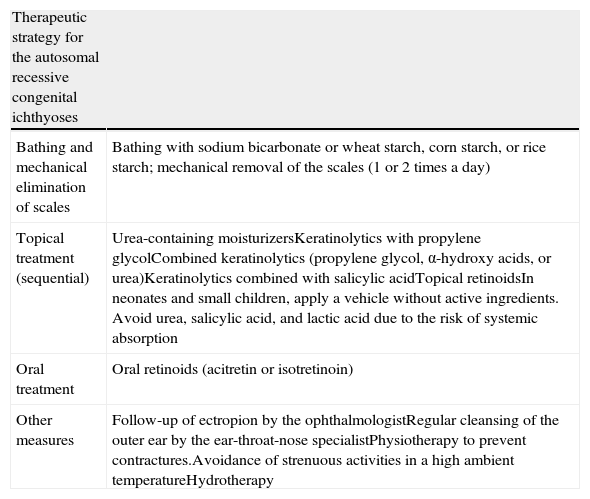
TreatmentThe primary aim of treatment in ichthyosis is to eliminate scaling and reduce xerosis without causing excessive irritation (Table 3). Before deciding on treatment, aspects such as age and sex of the patient, type and severity of the disease, and extent and site of the lesions should be taken into consideration.128
Therapeutic Strategy in Autosomal Recessive Congenital Ichthyoses.
| Therapeutic strategy for the autosomal recessive congenital ichthyoses | |
| Bathing and mechanical elimination of scales | Bathing with sodium bicarbonate or wheat starch, corn starch, or rice starch; mechanical removal of the scales (1 or 2 times a day) |
| Topical treatment (sequential) | Urea-containing moisturizersKeratinolytics with propylene glycolCombined keratinolytics (propylene glycol, α-hydroxy acids, or urea)Keratinolytics combined with salicylic acidTopical retinoidsIn neonates and small children, apply a vehicle without active ingredients. Avoid urea, salicylic acid, and lactic acid due to the risk of systemic absorption |
| Oral treatment | Oral retinoids (acitretin or isotretinoin) |
| Other measures | Follow-up of ectropion by the ophthalmologistRegular cleansing of the outer ear by the ear-throat-nose specialistPhysiotherapy to prevent contractures.Avoidance of strenuous activities in a high ambient temperatureHydrotherapy |
Daily bathing is recommended for patients with ARCI to mechanically eliminate scales and traces of moisturizer. This is easier if the patient is immersed in water for 15 to 30minutes. Some authors recommend adding sodium bicarbonate to the bath to denaturalize the keratins and make the water alkaline, and so facilitate elimination of the scales.129 Other products that can be added include wheat starch, corn starch, or rice starch. Bathing oils are not appropriate as they may lead to occlusion with subsequent risk of bacterial proliferation and worsening of thermoregulation.
Topical TreatmentMoisturizers and topical keratolytic agents are usually the first therapeutic option. They improve skin barrier function and facilitate desquamation. Mild local adverse effects, such as transient pruritus, irritation, or stinging sensation may occur.
Sodium chloride, urea, vitamin E acetate, glycerol, and petroleum jelly can be used as moisturizers and lubricants. In patients with thick scaling and marked hyperkeratosis, 1 or more keratolytic agents, such as α-hydroxy acids (lactic and glycolic acid),130 salicylic acid, N-acetylcystein,131–133 urea (>5%),134 and propylene glycol, can be added. Modulators of keratinocyte differentiation are also used. These include topical retinoids (tretinoin, adapalene, tazarotene),135,136 calcipotriol,137 and dexpanthenol.Topical retinoids often cause irritation and small, very painful fissures.137 Moreover, there is a risk of absorption and teratogenicity in fertile women if they are used too extensively.138 To enhance the effectiveness of keratolytics and moisturizers, occlusive dressing may be applied in specific areas refractory to treatment.139 An additive or synergistic effect can also be attained by combining 2 or more keratolytic agents or moisturizers.140–142 Treatment should be optimized for each individual, given the highly variable nature of the condition and skin sensitivity and differences in response to each treatment. The optimization process can be helped by treating one side of the body differently to the other to enable comparisons. Neonates and small children should be treated with a vehicle without any active substances as the skin is very fine and sensitive and most keratolytics are not tolerated. In addition, the risk of percutaneous absorption of topical products such as urea, salicylic acid, and lactic acid is greater.143–145
Systemic TreatmentOral retinoids have keratolytic effects that help eliminate scales and prevent excessive hyperkeratosis. Both isotretinoin and aromatic retinoids (acitretin and etretinate) have proved effective in the treatment of ARCIs.128,146,147 Acitretin at a dose of 0.5 to 1mg/kg/d is the most widely used drug, especially in patients with LI.148 Patients with CIE may have a more complete response and at lower doses.
The main adverse effects are mucocutaneous disorders, teratogenicity, musculoskeletal disorders, and abnormal lipid profile and transaminase elevation.149–152 With regards to teratogenicity, in the case of etretinate and acitretin, the drugs should be avoided during pregnancy and patients should avoid becoming pregnant for 3 years after discontinuation of treatment.151 Isotretinoin has a shorter half-life and is completely eliminated from the organism after 1 month and so may be the preferred option in women who wish to become pregnant.128
Treatment monitoring should include a laboratory work-up with a liver function test and lipid profile before starting treatment, then at 1 month and every 3 months after starting treatment. In fertile women, a pregnancy test should be performed in the 2 weeks before starting treatment and an effective contraceptive measure should be used from 4 weeks before treatment until 3 years afterwards (in the case of acitretin). When prolonged treatment is required with retinoids, growth and bone development should be monitored. Some authors suggest performing a bone study before treatment followed by a yearly examination.151 Recent guidelines do not recommend performing routine radiography because of the possible harmful effects.152 Instead, selective radiographic studies are recommended in patients who have atypical bone pain.152
An alternative to systemic retinoid treatment is the use of drugs known as retinoic acid metabolism blocking agents, which increase the endogenous levels of retinoic acid. One such drug is liarozole, which has been granted orphan status for the treatment of LI, CIE, and HI by the European Medicines Agency and the US Food and Drug Administration.153–155 This drug has been shown to be more effective than acitretin in clinical trials and it is also better tolerated and has a better pharmacokinetic profile.154
Other Medical CareIn patients with ectropion, the application of artificial tears and eye lubricants and moisturizing the skin of the face and the cheeks in particular can reduce palpebral retraction. Surgical correction is a valid option in severe cases, but this usually has to be repeated a few years later. Hydrotherapy may be beneficial.156 Patients should be advised to avoid strenuous physical activity when the ambient temperature is high, given that hypohidrosis carries with it the risk of heat stroke and convulsions. Oral retinoids can improve thermoregulation.157 Physiotherapy is important for preventing flexion contracture, particularly in the case of HI. Regular cleansing of the external auditory canal by an ear-throat-nose specialist can prevent scales from accumulating and so prevent hearing loss.
Genetic Counseling and Prenatal DiagnosisWhen a patient is diagnosed with ichthyosis, he or she should be offered appropriate genetic counseling in which the nature of the disorder, the transmission mode, and the risk of future manifestations in the family are explained. Prenatal diagnosis can indicate whether the fetus is affected and, if this is the case, psychological preparation of the family can be offered and problems anticipated during pregnancy and birth. The parents can be given the option of an abortion if no treatment is available. In addition, should gene therapy for these conditions become available in the future, prenatal diagnosis would enable application of this therapy as early as possible.
For more than 20 years, prenatal diagnosis was performed by taking a biopsy sample of fetal skin and studying it by optical microscopy, electron microscopy, or immunohistochemistry.158,159 This invasive procedure could only be performed in the late phases of pregnancy, between weeks 15 and 23 of gestation, and was associated with a 1% to 3% risk of losing the fetus.160,161 The identification of the molecular mechanisms of hereditary skin disorders has enabled a much earlier diagnosis based on genetic techniques.102,162–164 Fetal DNA is obtained by amniocentesis performed between weeks 15 and 20 or by chorionic villus sampling between weeks 10 and 12. The risk of fetal loss with these techniques is less than between 0.5% and 1%.165 Other noninvasive methods in development are analysis of fetal cell DNA and free fetal DNA in maternal circulation166 as well as the use of 3-dimensional ultrasound.167,168
Preimplantation genetic diagnosis could also be possible in in vitro fertilization techniques, such that only fertilized eggs free of the mutation are implanted in the uterus, thereby avoiding the need for abortion in most cases.169
Future Strategies for Genetic Treatment of IchthyosisAlthough important progress has been made in the genetic diagnosis of ichthyosis, new strategies are also being pursued for these diseases.170 The skin is the most accessible organ for gene transfer therapies, and so such techniques are minimally invasive.171 However, the skin also has unique immunologic characteristics that do not favor long-term expression of a transgenic product.172 In LI, a process of ex vivo gene transfer managed to restore normal TGM1 expression and correct the phenotype of skin transplanted on the back of immunosuppressed mice.173,174 Recently, the phenotype of cultured keratinocytes from patients with HI due to mutations in the ABCA12 gene has also been recovered.3
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Rodríguez-Pazos L, et al. Ictiosis congénitas autosómicas recesivas. Actas Dermosifiliogr. 2013;104:270–84.


