




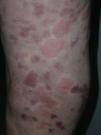
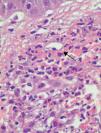



Urticarial vasculitis is a subtype of vasculitis characterized clinically by urticarial lesions and histologically by necrotizing vasculitis.
ObjectiveTo study the clinical and histologic features of urticarial vasculitis in patients seen in the dermatology department of Hospital Universitario Virgen de Rocío in Seville, Spain, and to examine the association between hypocomplementemia and systemic disease.
Material and methodsWe performed a chart review of histologically confirmed cases of urticarial vasculitis in the database of our department covering a period of 10 years.
ResultsFifteen patients (9 women and 6 men with a median age of 51 years) were included. In 14 patients (93%), the lesions persisted for more than 24hours, and in 9 cases (60%) the lesions resolved leaving residual purpura or hyperpigmentation. Seven patients (47%) had low complement levels in the blood, 12 (80%) had extracutaneous symptoms, and 8 (53%) had associated systemic disease, the most common of which was systemic lupus erythematosus.
ConclusionsUrticarial vasculitis may be underdiagnosed. Response to treatment is variable, and hypocomplementemia and extracutaneous symptoms may indicate the presence of associated systemic disease.
La urticaria vasculitis (UV) es un subtipo de vasculitis caracterizada clínicamente por lesiones de tipo urticarial e histológicamente por vasculitis necrotizante.
ObjetivoEl objetivo de este trabajo es el estudio de los pacientes con urticaria vasculitis que han sido atendidos en el Servicio de Dermatología del Hospital Universitario Virgen del Rocío (Sevilla, España) para determinar las características de presentación clínicas e histológicas, así como el despistaje de hipocomplementemia, en estrecha relación con enfermedad sistémica asociada.
Material y métodosSe han extraído de la base de datos de nuestro servicio los casos de urticaria vasculitis confirmados histológicamente durante un período de 10 años.
ResultadosQuince pacientes fueron diagnosticados de urticaria vasculitis (9 mujeres y 6 hombres, con una mediana de edad de 51 años). En 14 pacientes (93%) las lesiones persistían más de 24 horas. En 9 pacientes (60%) las lesiones se resolvían con púrpura e hiperpigmentación residual. Siete pacientes (47%) presentaron niveles disminuidos de complemento en sangre, 12 pacientes (80%) asociaron síntomas extracutáneos y 8 pacientes (53%) asociaban enfermedad sistémica, siendo la entidad más frecuente el lupus eritematoso sistémico.
ConclusionesLa urticaria vasculitis constituye a menudo una entidad infradiagnosticada, con una variable respuesta al tratamiento y en la que la existencia de niveles de complemento disminuidos y de síntomas extracutáneos puede ser indicativa de enfermedad sistémica asociada.
Urticarial vasculitis (UV) is a form of vasculitis that affects the small blood vessels and is characterized clinically by urticarial lesions and histologically by leukocytoclastic vasculitis. It may be idiopathic or associated with connective-tissue diseases (systemic lupus erythematosus [SLE] and Sjögren syndrome); neoplasms or associated conditions (lymphoma, monoclonal gammopathy); chronic infections with hepatitis viruses B and C, Epstein-Barr virus, or Borrelia burgdorferi; serum sickness; drugs (cimetidine, fluoxetine, procainamide, atenolol, sulfamethoxazole, paroxetine, sodium valproate, ciprofloxacin); and sun exposure.1 Patients with idiopathic UV may have normal or low complement levels, but systemic involvement is more common in the second case. UV is a major criterion for hypocomplementemic urticarial vasculitis syndrome (HUVS), a rare autoimmune disease with well-defined anatomic and clinical criteria that occurs in 5% of patients with chronic urticaria.2
UV is more common in adults than in children (in whom it is rare) and its incidence peaks in the fourth decade of life.2 It is also more common in women (60%-80% of cases).
Its pathogenesis is thought to be mediated by a type III hypersensitivity reaction, with the formation of immune complexes that can be endogenous (resulting from the production of immunoglobulin [Ig] G autoantibodies) or exogenous (induced by drugs or certain viral infections). The target of these autoantibodies is found in the C1q subunit of complement component C1. Between 30% and 75% of patients with UV have circulating immune complexes, which activate the complement system via the classical pathway.
Clinically, UV is characterized by urticarial plaques that last for over 24hours and resolve leaving purpura or hyperpigmentation; the lesions may also cause pain or a burning sensation. Patients can develop angioedema, arthralgia, abdominal or chest pain, fever, lung or renal disease, episcleritis, or uveitis. Systemic manifestations are more common in hypocomplementemic UV, in forms of UV associated with systemic disease (SLE, mixed connective tissue disease, rheumatoid arthritis, familial Mediterranean family fever, juvenile chronic arthritis, etc.), and in HUVS.1,2
The aim of this study was to describe the clinical and histologic features of UV in patients seen at the dermatology department of our hospital and to study blood complement levels, the presence of associated systemic disease, and the treatments used.
Patients and MethodsPatientsWe studied patients with UV seen at the dermatology department of Hospital Universitario Virgen del Rocio in Seville, Spain between January 1, 1998 and December 31, 2008; the criteria for inclusion were the presence of urticarial lesions with histologic features of leukocytoclastic vasculitis.
MethodsWe performed a retrospective, descriptive study of the epidemiological and clinical features of UV and the treatments administered based on information collected from patient charts and laboratory and biopsy records. Hypocomplementemia was defined as C3 levels of less than 90mg/dL (normal, 90-80mg/dL), C4 levels of less than 10mg/dL (normal, 10-40mg/dL), and C1q levels of less than 10mg/dL (normal, 10-25mg/dL). The study variables were age, sex, clinical characteristics, blood complement levels, antinuclear antibody (ANA) levels, presence of associated systemic disease, and treatment. A descriptive analysis of the study variables was performed and qualitative variables were reported using absolute and relative frequencies. Quantitative variables were expressed as median values (interquartile range [IQR]). The Fisher exact test was used to test for significant differences (P<.05) in qualitative variables between patients with normal and low complement levels.
ResultsFifteen patients (60% women and 40% men) were included in the study. The median age at diagnosis was 51 years (IQR, 23-56 years) and the median duration of disease was 24 months (IQR, 12-60 months). Table 1 summarizes patient data, complement levels, and associated systemic disease.
Patient Data, Blood Complement Levels, and Associated Systemic Diseases in Patients With Urticarial Vasculitis.
| Patient | Age, y | Sex | Complement Levels | Associated Systemic Disease |
| 1 | 53 | F | Low | Hypocomplementemic urticarial vasculitis syndrome |
| 2 | 69 | F | Low | Rheumatoid arthritis |
| 3 | 23 | F | Low | Mixed connective-tissue disease |
| 4 | 50 | F | Low | |
| 5 | 66 | F | Low | Systemic lupus erythematosus |
| 6 | 6 | F | Low | Familial Mediterranean fever |
| 7 | 8 | M | Normal | Juvenile chronic arthritis |
| 8 | 52 | M | Normal | Systemic lupus erythematosus |
| 9 | 47 | F | Low | None |
| 10 | 80 | F | Normal | None |
| 11 | 33 | F | Normal | None |
| 12 | 51 | M | Normal | None |
| 13 | 56 | M | Normal | None |
| 14 | 52 | M | Normal | None |
| 15 | 21 | M | Normal | None |
Abbreviations: F, female; M, male.
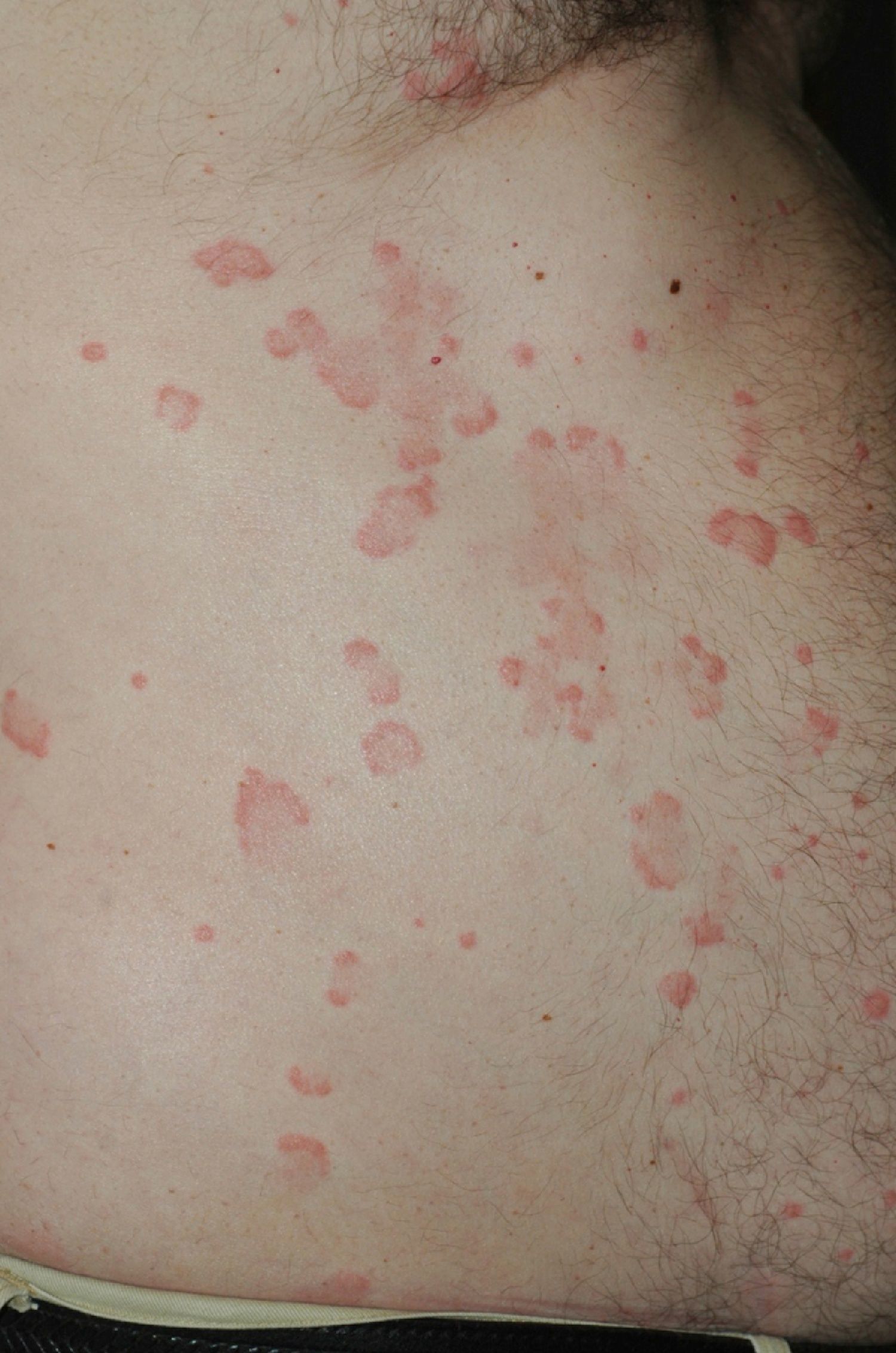
On clinical examination, all the patients had urticarial plaques, located mostly on the trunk and the proximal extremities (Figs. 1 and 2).
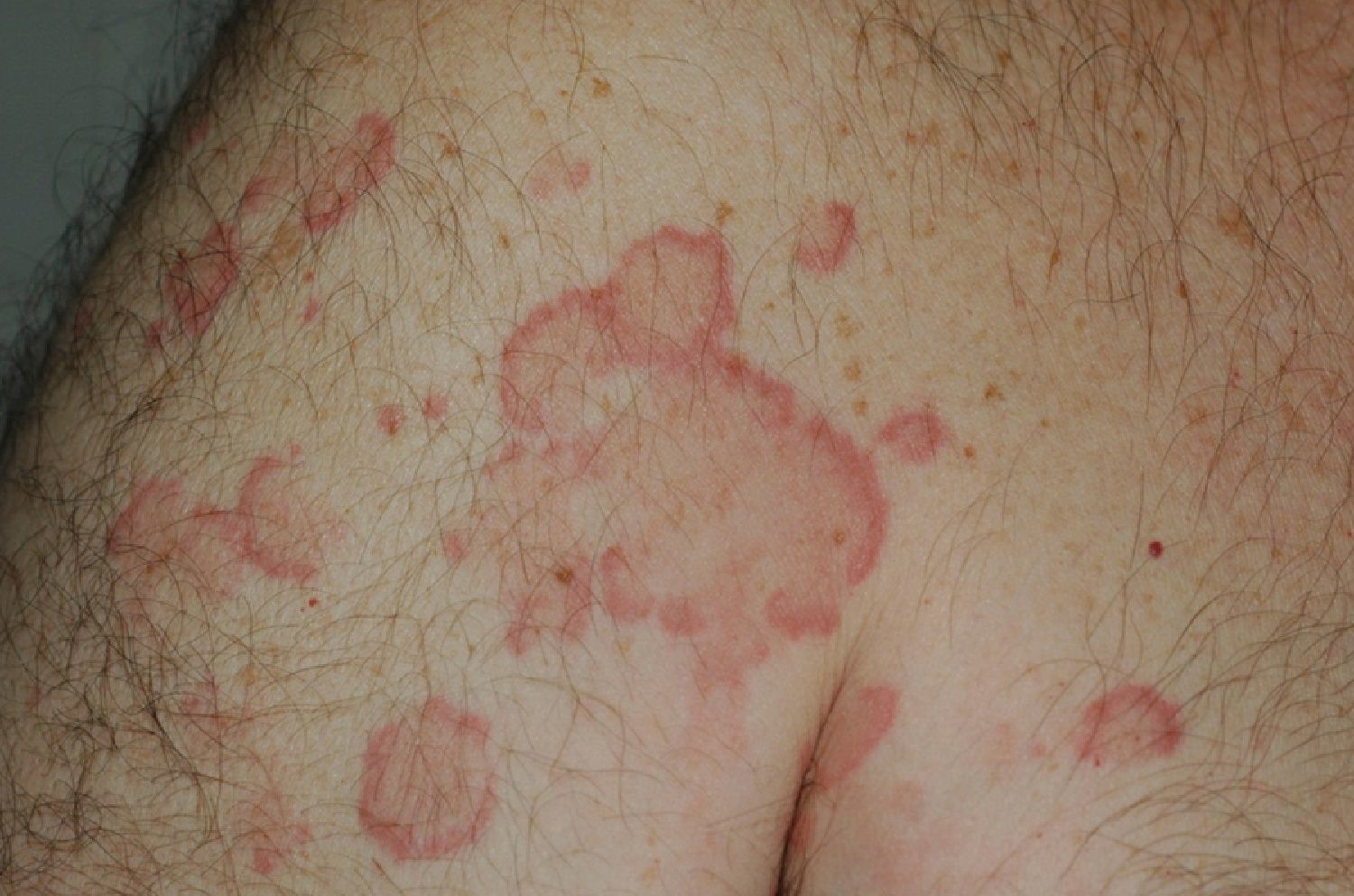
The lesions lasted for over 24hours in 14 patients (93%) including all 7 patients with low complement levels. They caused pain or a burning sensation in 2 patients (13%) and left residual hyperpigmentation or purpura on resolution in 9 (60%) (Fig. 3).
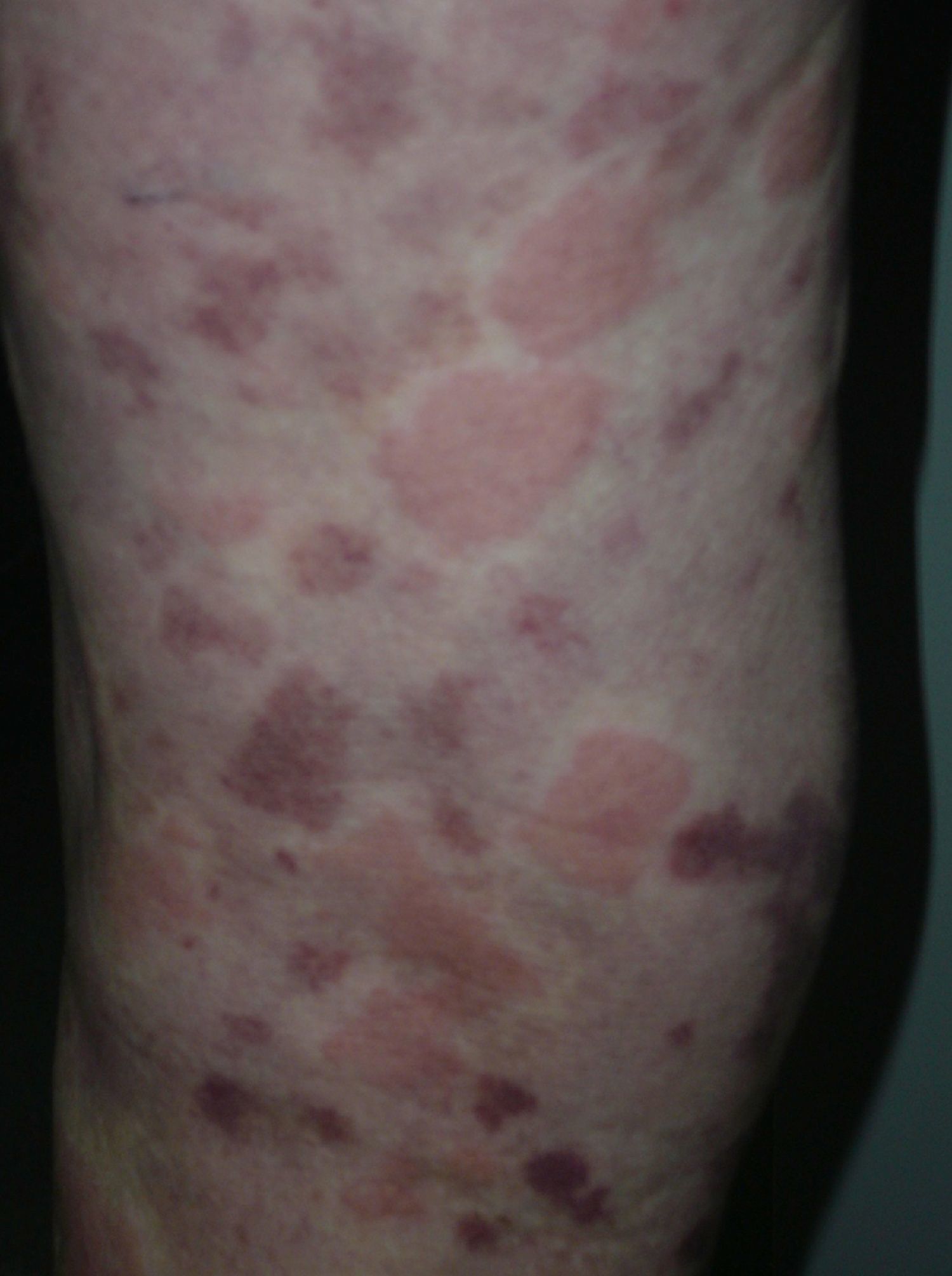
In 2 cases (13%), the lesions were accompanied by angioedema. Twelve patients (80%) had 1 or more extracutaneous symptoms, the most common of which were arthralgia and fever.
On analyzing the patients by complement levels, 8 (53%) had normal levels and 7 (47%) had low levels. Of the patients with hypocomplementemia, 4 (57.14%) had low C3, C4 and C1q levels, 2 (28.57%) had low C3 and C1q levels, and 1 (14.28%) had low C4 levels (Table 2).
Clinical Findings According to Complement Levels.
| Clinical Findings | Hypocomplementemia | Normocomplementemia | ||
| (n=7) | % | (n=7) | % | |
| Duration of lesions >24h | 7 | 100 | 7 | 87.5 |
| Duration of lesions <24h | - | - | 1 | 12.5 |
| Pain or burning sensation | 2 | 28.6 | - | - |
| Residual hyperpigmentation | 4 | 57.1 | 5 | 62.5 |
| Angioedema of the face or hands | 2 | 28.6 | - | - |
| Associated arthralgia at onset of disease | 2 | 28.6 | 2 | 25 |
| Obstructive pulmonary disease | 1 | 14.3 | - | - |
| Abdominal pain | 1 | 14.3 | - | - |
| Fever | 4 | 57.1 | 2 | 25 |
| Immunoglobulin or C3 deposits | 2 | 28.6 | 1 | 12.5 |
| Vessel walls | - | - | - | - |
| Basement membrane | 2 | 28.6 | 1 | 12.5 |
All 7 of the patients with low complement levels were women and 6 of them (88%) had associated systemic disease. The 2 cases of angioedema occurred in patients with hypocomplementemia; 1 had HUVS and the other had mixed connective-tissue disease.
ANA positivity was observed in 5 patients (33%), all of whom had an associated systemic condition. The same 5 patients also had low complement levels, demonstrating a significant association between ANA positivity and hypocomplementemia (P=.002).
All 15 patients had histologic findings consistent with leukocytoclastic vasculitis (Fig. 4).
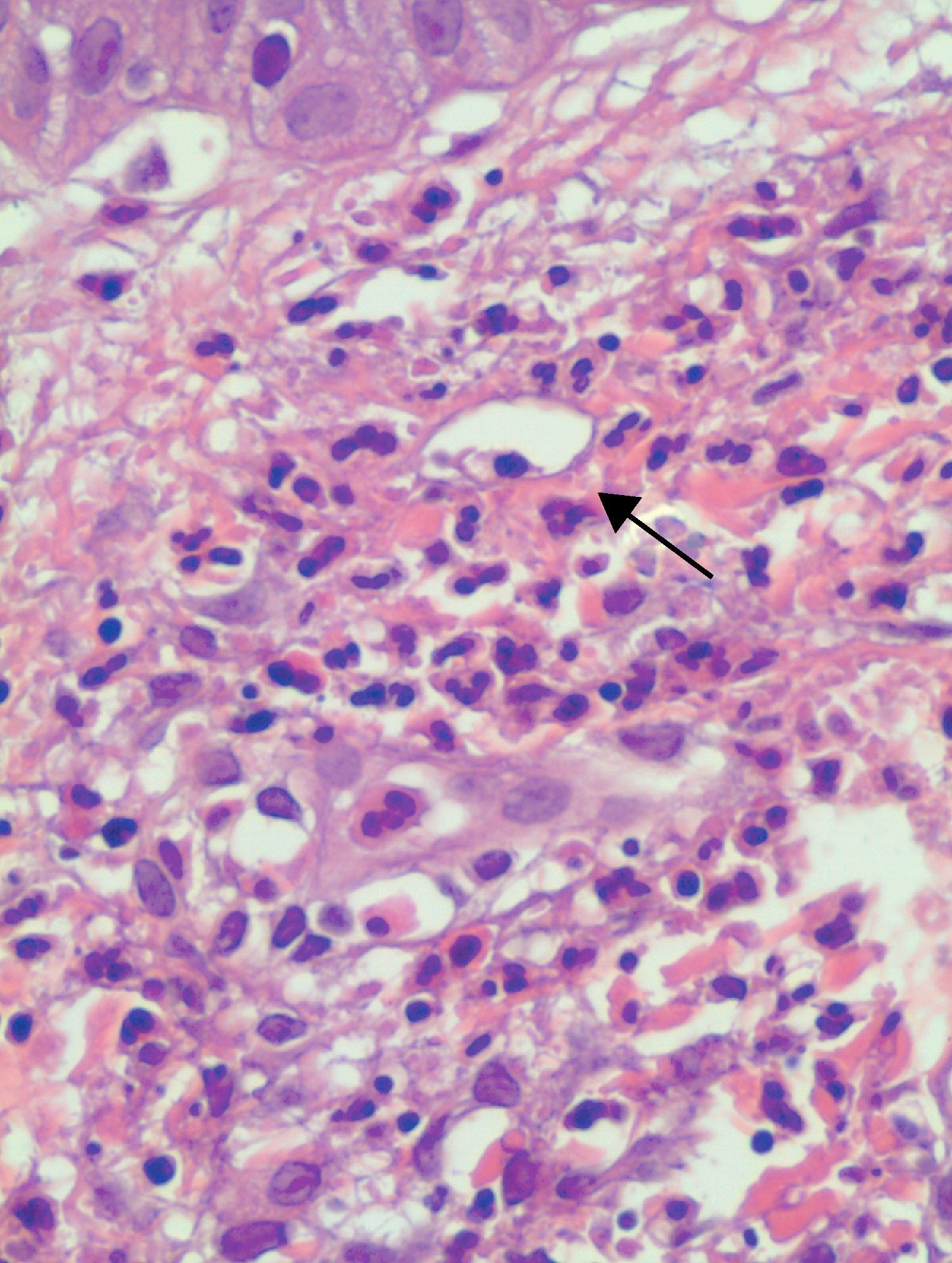
Three (37.5%) of the 8 patients in whom a direct immunofluorescence study was performed (53%) had a positive result. Of these, 2 had granular deposits of C3 at the dermal-epidermal junction, in both cases associated with hypocomplementemia and systemic disease (SLE in one case and mixed connective-tissue disease in the other). The third patient had linear IgM deposits at the dermal-epidermal junction. Direct immunofluorescence was negative in the remaining cases studied.
Seven patients (46.66%) had UV as the sole manifestation of disease and 8 (53%) had associated systemic involvement. The systemic disease preceded UV in 1 case and developed at the same time in the other 7. The most common associated disease was SLE (3 cases, 37.5%); the other diseases (present in 1 patient each, 12.5%) were mixed connective-tissue disease, rheumatoid arthritis, familial Mediterranean fever, juvenile chronic arthritis, and HUVS.
Table 3 shows the drugs used to treat UV in our series. Eight patients (53.33%) responded to a combination of antihistamines and oral corticosteroids, 2 (13.33%) to colchicine, 1 (6.66%) to indomethacin, and 1 (6.66%) to dapsone. The remaining 3 patients (20%) were receiving rituximab or cyclophosphamide infusions to treat the underlying disease.
DiscussionUV is a rare but possibly underdiagnosed disease. The clinical presentation ranges from cutaneous manifestations alone to skin disease with associated systemic disease; the presence of an underlying systemic disease is more common in hypocomplementemic UV. Since our hospital has a catchment population of approximately 600 000, the fact that only 15 cases of UV were recorded over a 10-year period would, based on our experience, appear to support the hypothesis that UV is an underdiagnosed condition.1–4 In a study of 47 patients with UV, Tosoni et al.1 noted that not all patients fulfilled the classical diagnostic clinical criteria, a finding that could explain why UV is underdiagnosed. Under half of the patients had lesions that lasted for more than 24hours, under 9% reported a burning sensation, and only 6% had residual purpura or hyperpigmentation after resolution of the lesions. On the basis of their findings, those authors suggested that the duration and individual characteristics of lesions should not be considered major criteria as they are prone to subjective interpretation, and proposed instead that all chronic urticarial lesions refractory to treatment with antihistamines should be biopsied. Our findings are in agreement with most studies conducted to date in that practically all patients (93%) had lesions that lasted for longer than 24hours and left residual purpura or hyperpigmentation.5,6
Over half of the patients in our series had associated systemic involvement. This percentage is much higher than figures reported in other studies5,6 and it is possible that the difference is largely due to the fact that 7 of the 8 patients in our series had already been diagnosed with a systemic condition when they developed UV. The presence of systemic disease may have increased the index of suspicion of UV when these patients developed urticaria-like lesions. It is of note that one of the children in our series had familial Mediterranean fever (V726A mutation in the MEFV gene); we found no such associations in our review of the literature.7–9
UV can also form part of HUVS, a syndrome first described by McDuffie et al.10 in 1973 for which Schwartz et al.11 established major and minor criteria some years later. Extracutaneous manifestations must be present for a diagnosis of HUVS to be made; this is not the case with hypocomplementemic UV, in which patients may have cutaneous involvement alone or both cutaneous and extracutaneous involvement. Only 106 cases of HUVS have been published since the syndrome was first characterized, and 100 of these were in adults.12,13 Of note in our series was a female patient who fulfilled both the major and minor criteria for this rare syndrome.
There were more women than men in our series; this finding is consistent with previous reports, which have additionally noted that hypocomplementemic UV almost exclusively affects women.1,2,5 In our study, all the patients with low serum complement levels were women.
UV tends to develop in patients aged between 40 and 50 years. Of note was the fact that our series included 2 children, as pediatric cases were rare in the studies we reviewed.7,12,13 Both of the children had associated systemic disease (familial Mediterranean fever in one case and chronic juvenile arthritis in the other).
Eighty percent of the patients in our series had one or more extracutaneous symptoms, the most common being fever (40%) and arthralgia (27%); these findings coincide with reports from the larger series we analyzed.1,5,6 It is interesting to note that the patients with low complement levels had a greater tendency than those with normal levels to experience pain, a burning sensation, or systematic symptoms such as fever and arthralgia. Other authors have also noted this higher prevalence of extracutaneous symptoms in patients with hypocomplementemic UV.1,5,6
Histologically, UV is characterized by features consistent with leukocytoclastic vasculitis. Biopsy of active lesions is the diagnostic test of choice for UV, and histologic evidence of leukocytoclastic vasculitis is found in most cases.14,15 In 70% of cases, direct immunofluorescence findings include linear or granular deposits of immunoglobulins, complement, or fibrin in the area of the basement membrane or the vascular endothelium.16–18 In our series, only 3 (37%) of the 8 patients for whom direct immunofluorescence results were available had deposits at the dermal-epidermal junction; of these, 2 had associated systemic involvement and low complement levels. This last finding is consistent with reports by other authors that patients with positive direct immunofluorescence results tend to have low blood complement levels.5,6
Complement levels are very important in UV. Seven (47%) of the patients in our series had low complement levels and, as already mentioned, this proportion is higher than that reported in studies of larger samples.1,5,6,18 The most commonly affected complement components are C1 (C1q), C2, C3, and C4, which are all components of the classical pathway. Several authors have stated that hypocomplementemia is a marker of systemic disease.1,5,6 In our study, 53% of patients had autoimmune systemic disease, which is associated with an increased risk of hypocomplementemia. In fact, 6 (88%) of the 7 patients with low complement levels had associated systemic involvement; this percentage is much higher than figures reported by Mehregan et al.5 and Dincy et al.6 The probable reason is that most of the patients in our series had been previously diagnosed with an underlying systemic disease in which complement levels are typically low. SLE is among the systemic diseases most frequently associated with UV. In fact, the overlapping of symptoms between UV and SLE has led some authors to postulate that they might be variants of the same disease.19,20
UV is difficult to treat and treatment is often dictated by the severity of symptoms and by the underlying systemic disease. Antihistamines are the first-line drug for the treatment of UV with skin involvement only. Cinnarizine, which has antihistamine, vasoactive, and complement inhibitory properties, has also been used.2 Antihistamines, however, do not control the inflammation caused by immune complexes; in such cases, other treatments are required, such as oral corticosteroids, indomethacin, colchicine, dapsone, and hydroxychloroquine; of these, corticosteroids achieve symptomatic relief in the largest proportion of patients.2,5 In several cases, as shown by our results, patients may require treatment with immunosuppressants such as cyclophosphamide or azathioprine, or with drugs such as rituximab or intravenous IgG.12,21 Such treatment is mainly necessary in patients who have an associated systemic condition.
ConclusionsThe clinical characteristics of UV in our series are similar to those described in previous studies, with lesions that tended to last for more than 24hours and leave residual pigmentation on resolution. Of note in our series was the high percentage of hypocomplementemic patients with associated systemic disease. This highlights the importance of measuring complement levels in patients with UV, as low levels tend to be associated with a higher frequency of extracutaneous symptoms and are therefore an indication of underlying systemic disease. Finally, the presence of an autoimmune systemic disease should raise suspicion of UV in patients with urticaria-like lesions.
Ethical DisclosuresProtection of Humans and AnimalsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of DataThe authors declare that they have followed the protocols of their work center on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to Privacy and Informed ConsentThe authors declare that no private patient data appear in this article.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Moreno-Suárez F., et al. Urticaria vasculitis: estudio retrospectivo de 15 casos. Actas Dermosifiliogr. 2013;104:579–85.


