


Una niña recién nacida fue valorada por nuestro Servicio de Dermatología por una lesión tumoral congénita en el área frontal derecha. Era la primera hija de padres sanos no consanguíneos, nacida a término tras un embarazo y parto normales. No había antecedentes maternos de infecciones o ingesta de drogas ni antecedentes familiares de enfermedades cutáneas.
Exploración físicaSe observaba un nódulo eritematoso de aspecto vascular, ulcerado, de consistencia fibrosa, adherido a planos profundos y de aproximadamente 3cm de diámetro (fig. 1). No presentaba otras lesiones cutáneas similares ni se palpaban visceromegalias.
Histología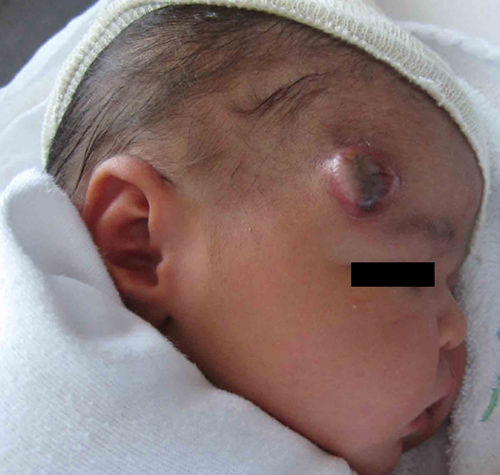
La histología mostraba, en la dermis superficial y profunda, una proliferación de células fusiformes sin atipia nuclear, que se agrupaban en bandas y fascículos (figs. 2 A y B). Con técnicas de inmunohistoquímica se demostró positividad para vimentina y actina alfa de músculo liso (fig. 2 C) y negatividad para S-100, mioglobina, citoqueratinas y desmina (fig. 2 D).
Exploraciones complementarias
La serie ósea, ecografía abdominal y tomografía axial computarizada no permitieron detectar afectación ósea ni visceral.
¿Cuál es su diagnóstico?
DiagnósticoMiofibromatosis infantil solitaria.
Evolución y tratamientoSe decidió mantener actitud expectante con revisiones periódicas. El tumor sigue regresando progresivamente.
ComentarioLa miofibromatosis infantil (MI) es una enfermedad mesenquimal congénita caracterizada por la presencia de tumores únicos o múltiples de origen miofibroblástico que puede afectar la piel, los tejidos blandos, el hueso y los órganos internos1. Esta entidad fue descrita por Stout en 1954, pero el término actual de MI fue introducido en 1981 por Chung y Enzinger2. Aunque se considera una enfermedad rara, la MI constituye el tumor fibroso más frecuente en la infancia. Habitualmente se manifiestan desde el nacimiento hasta los 2 años de vida y las lesiones que se limitan a la piel suelen tener buen pronóstico, con altas tasas de regresión espontánea.
La mayoría de los casos de MI son esporádicos, aunque se han descrito casos familiares en gemelos y en generaciones sucesivas, lo cual podría explicarse por un patrón de herencia autosómico dominante con penetrancia variable3.
La etiología es desconocida. Yousefi et al.4 propusieron que las células madre mesenquimales transferidas durante el embarazo podrían constituir el tejido tumoral en el feto, pero tras estudiar muestras de tejido tumoral de 4 recién nacidos con MI solitario o múltiple demostraron que las células tumorales no derivaban de células maternas quiméricas. El nivel de estrógenos maternos sí parece tener influencia sobre el desarrollo de la MI, debido a la regresión tumoral espontánea después del nacimiento, cuando la exposición a estrógenos desaparece1,5.
Existen 3 patrones clínicos de presentación: MI solitaria (una lesión única afectando la piel y/o el músculo de la cabeza, el cuello o el tronco, más común en niños y que representa el 75% de todos los casos); MI multicéntrica sin afectación visceral (lesiones múltiples limitadas a la piel y el músculo) y MI multicétrica con afectación visceral o generalizada (lesiones múltiples no solamente en la piel y/o el músculo, sino también en el hueso, el pulmón, el corazón y el tracto gastrointestinal)1. Las 2 últimas formas corresponden al 25% de todos los casos y son más frecuentes en niñas.
La morfología clínica de los tumores cutáneos de MI es heterogénea; son placas, nódulos o masas, solitarias o múltiples, de 0,5 a 5cm, no dolorosas, de consistencia firme, que rara vez se ulceran o sangran y que pueden tener un aspecto queloideo o vascular.
La biopsia de las lesiones accesibles (habitualmente las cutáneas) es necesaria para el diagnóstico de confirmación. En los tumores cutáneos de MI se observa un nódulo dérmico bien delimitado con una apariencia bifásica. En la periferia se encuentran abundantes células fusiformes agrupadas en fascículos (fascículos músculo liso-like); estas células no tienen atipias nucleares, aunque pueden presentar mitosis ocasionales, y expresan actina alfa del músculo liso y vimentina, siendo negativas para S100. En la zona central se observan estructuras vasculares con luces irregulares y patrón hemangiopericitoide6.
El diagnóstico diferencial en el caso de lesiones aisladas se establece con hemangiomas profundos, neurofibromas, leiomiomas, sarcomas y metástasis de neuroblastoma. Desde un punto de vista histopatológico debemos diferenciarlo del fibrosarcoma congénito y hemangiopericitoma3,5.
El pronóstico de las lesiones solitarias y múltiples sin afectación visceral es excelente, con regresión espontánea en 1-2 años secundaria probablemente a la apoptosis masiva1. Ante estos tipos de MI debe plantearse una actitud conservadora y expectante. Por el contrario, la MI con afectación visceral es una enfermedad grave, sobre todo por el compromiso gastrointestinal y cardiopulmonar, con una mortalidad elevada6, por lo que estos pacientes requerirán tratamiento quirúrgico y/o médico (como radioterapia o quimioterapia con vincristina, actinomicina D y ciclofosfamida) junto a cuidados paliativos.


