







La micosis fungoide (MF) es el linfoma cutáneo primario de células T más frecuente. Su aparición en la infancia es excepcional.
ObjetivosDescribir las características epidemiológicas, clínicas, histopatológicas e inmunofenotípicas de los pacientes con MF. Describir los tratamientos utilizados y la evolución.
Material y métodoSe incluyeron todos los pacientes admitidos en el Hospital de Pediatría Dr. J. P. Garrahan (Argentina) en el período comprendido entre agosto de 1988 y julio de 2014 con diagnóstico clínico e histopatológico de MF.
ResultadosSe diagnosticaron 14 pacientes con MF. La distribución por sexo fue M/F: 1:1,33. La edad media al diagnóstico fue de 11,23 años (rango: 8 a 15 años). El tiempo promedio de evolución hasta el momento del diagnóstico fue de 3 años y 6 meses (rango: 4 meses a 7 años). Todos los pacientes presentaron la forma clínica hipopigmentada y en el 42% se asoció la forma clásica. El 50% (n=7) exhibió un inmunofenotipo CD8 positivo de forma exclusiva. El 78% presentó estadio IB. La fototerapia fue el tratamiento de elección. Cuatro pacientes tuvieron por lo menos una recaída y 3 demostraron progresión de su enfermedad a nivel cutáneo. La evolución fue favorable en todos los casos.
ConclusionesLa MF es una entidad infrecuente en la infancia, siendo la forma hipopigmentada la más frecuente. Su diagnóstico es tardío debido a la similitud con otras enfermedades hipopigmentadas frecuentes en la niñez. A pesar de tener un buen pronóstico, presenta alta tasa de recidivas y requiere un seguimiento a largo plazo.
Mycosis fungoides (MF), the most common primary cutaneous T-cell lymphoma, is unusual in children.
ObjectivesWe aimed to describe the epidemiologic, clinical, histopathologic, and immunophenotypic characteristics of MF as well as treatments and course of disease in a pediatric case series.
Material and methodData for all patients admitted to our pediatric hospital (Hospital Dr. J. P. Garrahan) in Argentina with a clinical and histopathologic diagnosis of MF between August 1988 and July 2014 were included.
ResultsA total of 14 patients were diagnosed with MF. The ratio of boys to girls was 1:1.33. The mean age at diagnosis was 11.23 years (range, 8–15 years). The mean time between onset and diagnosis was 3.5 years (range, 4 months–7 years). All patients had hypopigmented MF and 42% also presented the features of classic MF. Seven (50%) had the CD8+ immunophenotype exclusively. Seventy-eight percent were in stage IB at presentation. Phototherapy was the treatment of choice. Four patients relapsed at least once and skin lesions progressed in 3 patients. All patients improved.
ConclusionsMF is unusual in children. The hypopigmented form is the most common. Diagnosis is delayed because the condition is similar to other hypopigmented diseases seen more often in childhood. Although prognosis is good, the rate of recurrence is high, so long-term follow-up is necessary.
La micosis fungoide (MF) es el linfoma cutáneo primario de células T más frecuente y afecta predominantemente a personas adultas en la quinta década de la vida1. Su manifestación clínica clásica presenta 3 fases evolutivas (mácula, placa y tumor) que pueden superponerse. Es habitual que haya un compromiso cutáneo exclusivo durante años o décadas, con un curso indolente. El epidermotropismo del infiltrado tumoral y la proliferación de linfocitos T atípicos de aspecto cerebriforme son los hallazgos histopatológicos característicos.
En la infancia la presencia de linfomas cutáneos primarios (LCP) es excepcional. La MF es poco habitual en niños, sin embargo, al igual que sucede en la población adulta, es el LCP más frecuente2. Las primeras descripciones de MF en niños datan del año 19843. Desde entonces existen diferentes publicaciones, en la mayoría de las cuales la variedad clínica hipopigmentada es la más habitual2,4–11. La histopatología no muestra diferencias con la MF del adulto, pero en los estudios inmunohistoquímicos predomina el infiltrado de linfocitos T CD8+, lo cual se correlacionaría con la forma clínica hipocrómica.
Los objetivos del presente trabajo son describir las características epidemiológicas, clínicas, histopatológicas, inmunofenotípicas y de reordenamiento clonal del receptor de linfocitos T (TCR) de los pacientes pediátricos con diagnóstico de MF, además de los tratamientos utilizados y de la evolución de la enfermedad.
Sujetos y métodoSe diseñó un estudio retrospectivo, observacional, descriptivo y transversal. En él se incluyeron los pacientes con diagnóstico clínico e histopatológico de MF atendidos entre agosto de 1988 y julio de 2014 en nuestro hospital (Hospital de Pediatría S.A.M.I.C. «Prof. Dr. Juan P. Garrahan» de Buenos Aires, centro de referencia nacional y de Latinoamérica). Las variables analizadas fueron las siguientes: sexo, edad, diagnóstico inicial, tiempo de evolución, manifestaciones clínicas, localización, estadio inicial, histopatología, inmunofenotipo, reordenamiento clonal del TCR, tratamientos recibidos, evolución y tiempo de seguimiento. Se realizó una revisión de las historias clínicas y de los estudios histopatológicos utilizando la clasificación de la World Health Organization/European Organization for Research and Treatment of Cancer del año 20051 para el diagnóstico; y la estadificación International Society for Cutaneous Lymphomas/European Organization for Research and Treatment of Cancer del año 200712.
ResultadosSe incluyeron 14 pacientes con criterios clínicos e histológicos de MF. La distribución por sexo fue M/F: 1:1,33. La edad media al diagnóstico fue de 11,23 años (rango: 8 a 15 años). El tiempo promedio de evolución hasta el momento del diagnóstico fue de 3 años y 6 meses (rango: 4 meses a 7 años). Los datos demográficos, características clínicas, diagnóstico histopatológico, inmunohistoquímica, reordenamiento clonal del TCR, el estadio, tratamiento, evolución, seguimiento y meses libres de enfermedad se detallan en las tablas 1 y 2.
Datos clínicos
| N° | S/E | TE | Clínica | Localización | Diagnóstico clínico inicial |
|---|---|---|---|---|---|
| 1 | F/10 | 4 | Placas y tumores MH y MC | Diseminada | MF |
| 2 | F/13 | 4 | Placas MH | Tronco Extremidades superiores | Dermatitis atópica |
| 3 | M/13 | 1 | MH | Tronco | MF |
| 4 | F/12 | 1,5 | Placas MH y MC | Abdomen Miembros inferiores | MF |
| 5 | F/9 | 5,1 | MH | Tronco y extremidades | Papulosis linfomatoide |
| 6 | F/14 | 2 | MH | Tronco Abdomen | Pitiriasis versicolor acromiante |
| 7 | F/11 | 0,5 | MH | Extremidades superiores | MF |
| 8 | M/13 | 0,3 | MH | Extremidades | Vitíligo |
| 9 | M/8 | 3 | Placas MH | Abdomen Extremidades | PL |
| 10 | F/10 | 3 | Placas MH | Tronco Extremidades | MF |
| 11 | M/10 | 6 | Placas MH | Tronco Extremidades | MF |
| 12 | M/8 | 7 | MH | Diseminada | PL |
| 13 | F/15 | 6 | MH | Diseminada | MF |
| 14 | M/13 | 6,1 | MH | Diseminada | MF |
MC: máculas clásicas; MF: micosis fungoide; MH: máculas hipopigmentadas; PL: pitiriasis liquenoide; S/E: sexo/edad al diagnóstico (en años); TE: tiempo de evolución (en años).
Datos clínicos, tratamiento y evolución
| N° | Diagnóstico HP e IHQ | TCR | TNM/Estadioa | Tratamiento | Evolución | Seguimiento | TLE |
|---|---|---|---|---|---|---|---|
| 1 | MF CD4– CD8+ | + Vb22 (94%) CD4 | T3 N3 M0/IVA2 | QT CHOP y electron beam | LE y recaída (1) | 7,51 años | 4 años |
| 2 | MF CD4– CD8+ | NR | T2b N0 M0/IB | – | PS | 3 meses | – |
| 3 | MF CD4+ CD8+ | - | T2b N0 M0/IB | PUVA UVB CC tópico | LE luego de recaída (1) | 3,5 años | 6 meses |
| 4 | MF CD4+ CD8– | + Gamma (piel) y Delta (sangre) | T2b N0 M0/IB | PUVA Bexaroteno tópico | En tratamiento luego de recaída (1) y progresión | 4 años | – |
| 5 | MF CD4+ CD8+ | NR | T2a N0 M0/IB | PUVA UVB Bexaroteno tópico | En tratamiento luego de recaídas (2) y progresión | 9 años | – |
| 6 | MF CD4+ CD8– | NR | T2a N0 M0/IB | PUVA CC tópico | En tratamiento | 9 meses | – |
| 7 | MF CD4+ CD8+ | NR | T1a N0 M0/IA | PUVA CC tópico | LE | 8 meses | Un mes |
| 8 | MF Folicular CD4– CD8+ | - | T1a N0 M0/IA | UVB CC tópico | LE | 13 meses | 3 meses |
| 9 | MF CD4- CD8+ | - | T2a N0 M0/IB | UVB+Psoraleno+retinoides CC tópico | Progresión En tratamiento | 13 meses | – |
| 10 | MF CD4– CD8+ | NR | T2a N0 M0/IB | UVB PUVA CC tópico | LE | 12 meses | Un mes |
| 11 | MF CD4– CD8+ | NR | T2a N0 M0/IB | PUVA | En tratamiento | 8 meses | – |
| 12 | MF CD4+ CD8+ | NR | T2a N0 M0/IB | UVA-1 CC tópico | En tratamiento | Un mes | – |
| 13 | MF CD4+ CD8+ | NR | T2a N0 M0/IB | PUVA CC tópico | En tratamiento | 2 meses | – |
| 14 | MF CD4+ CD8+ | + Beta | T2a N0 M0/IB | PUVA | En tratamiento | 2 meses | – |
CC: corticoide local; CHOP: ciclofosfamida, doxorrubicina, vincristina, prednisona; HP: histopatología; HT: helioterapia; IHQ: inmunohistoquímica; LE: libre de enfermedad; MF: micosis fungoide; NR: no realizado; PS: pérdida de seguimiento; QT: quimioterapia; TCR: rearreglo del receptor T; TLE: tiempo libre de enfermedad (número de recaídas).
Solo en 8 pacientes (57%) el diagnóstico inicial fue de MF. En los 6 pacientes restantes los diagnósticos iniciales fueron: pitiriasis liquenoide crónica (PLC) (2pacientes), papulosis linfomatoide (PL) (1 paciente), pitiriasis versicolor hipopigmentada (1 paciente), vitíligo (1 paciente) y dermatitis atópica (1 paciente) (tabla 1).
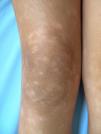
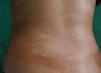
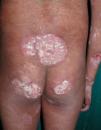
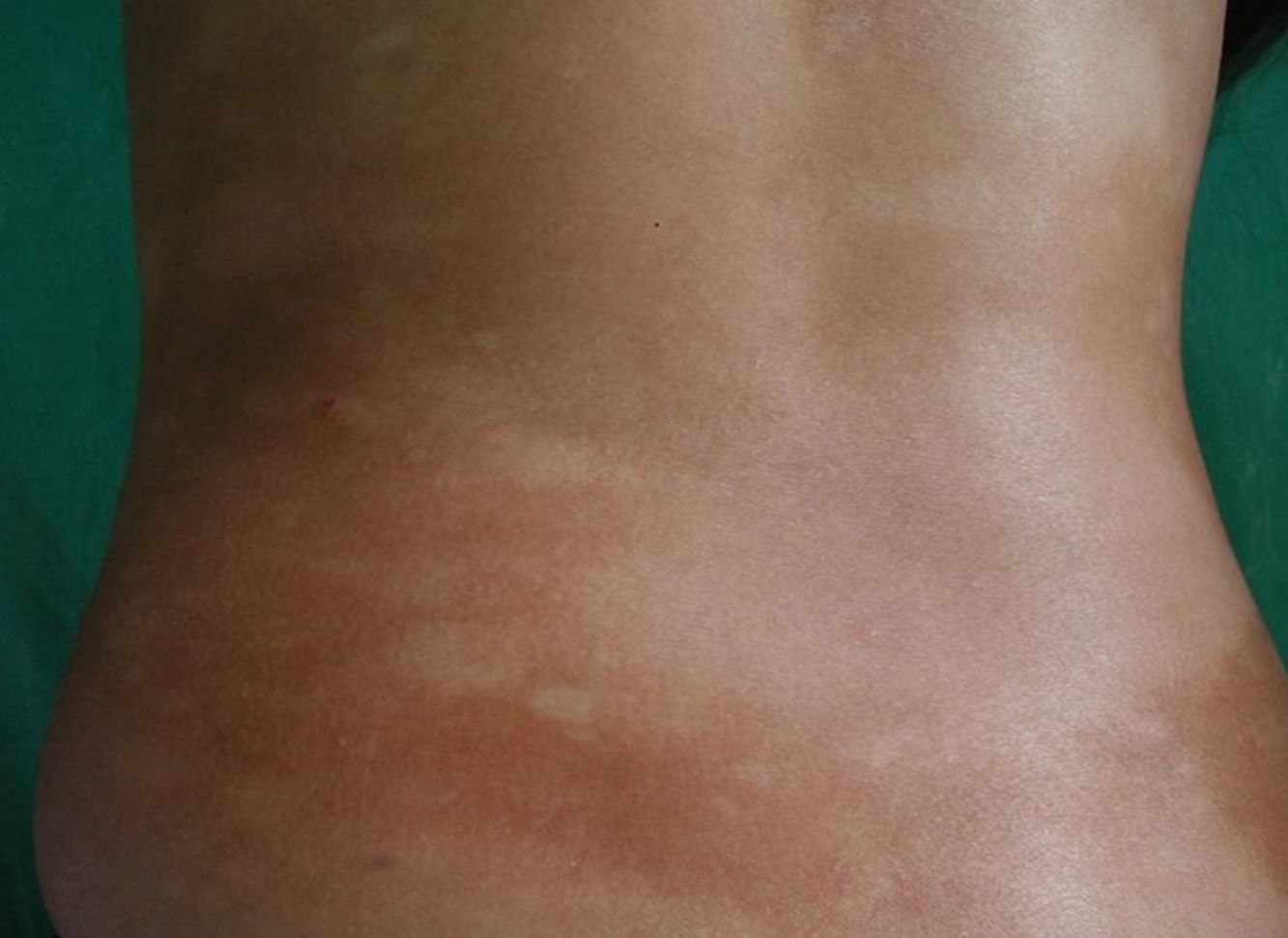
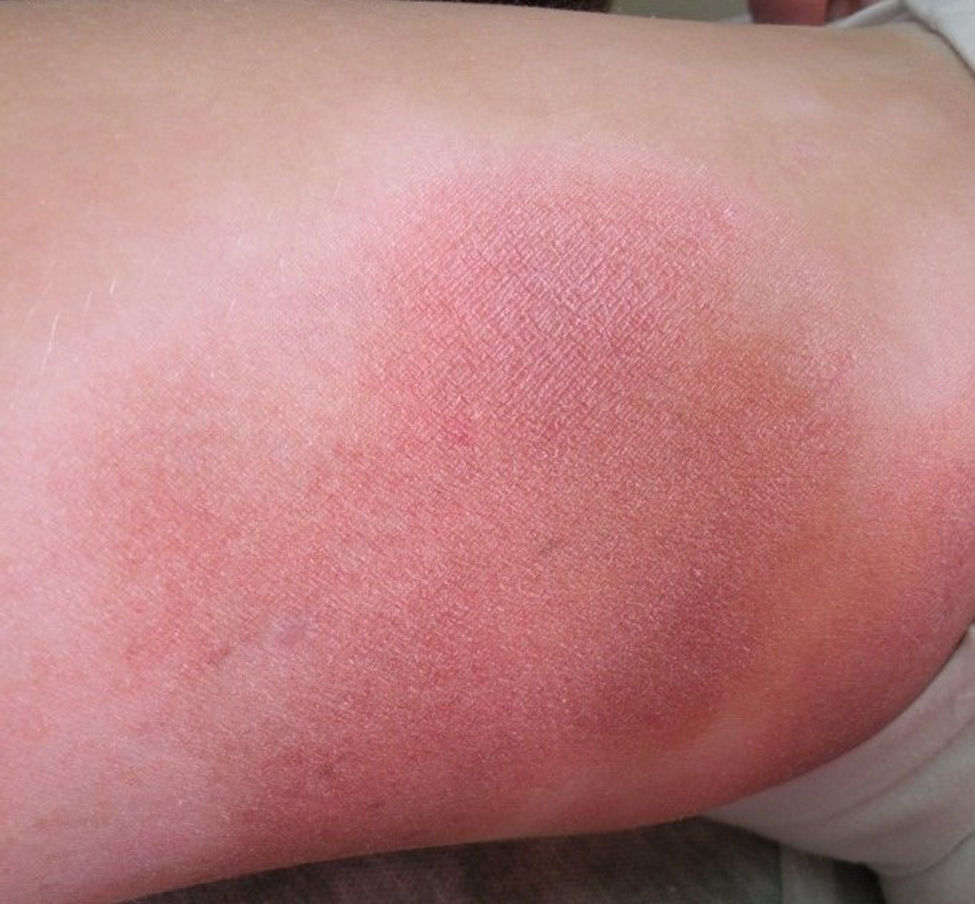
Respecto a las formas clínicas de la enfermedad, la totalidad de los pacientes presentó máculas hipopigmentadas (figs. 1 y 2). En 6 (42,8%) de ellos se observó, de forma concomitante o subsiguiente, la presencia de lesiones de MF clásica: máculas (2pacientes) (fig. 3), placas (6pacientes) (fig. 4) y tumores (1 paciente) (fig. 5). Además, un paciente (n.° 9) tuvo clínica de PLC, pero diagnóstico histopatológico de MF.
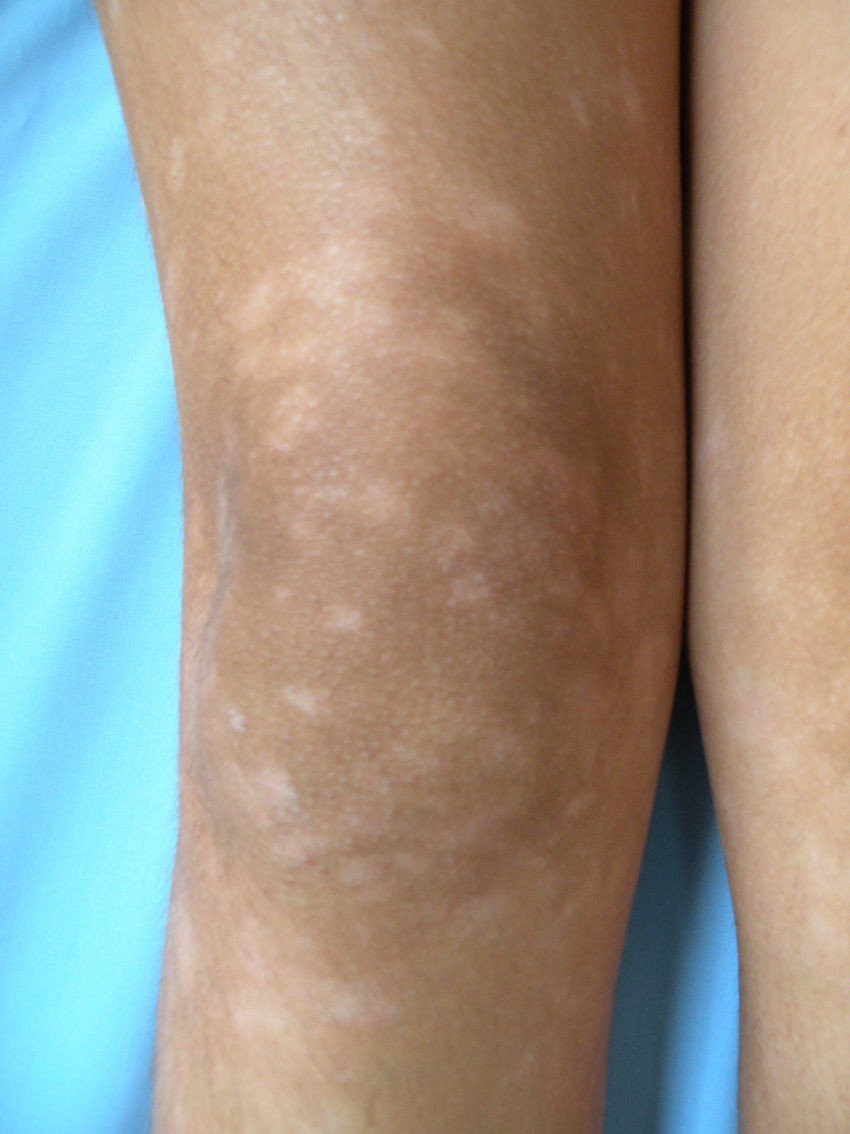
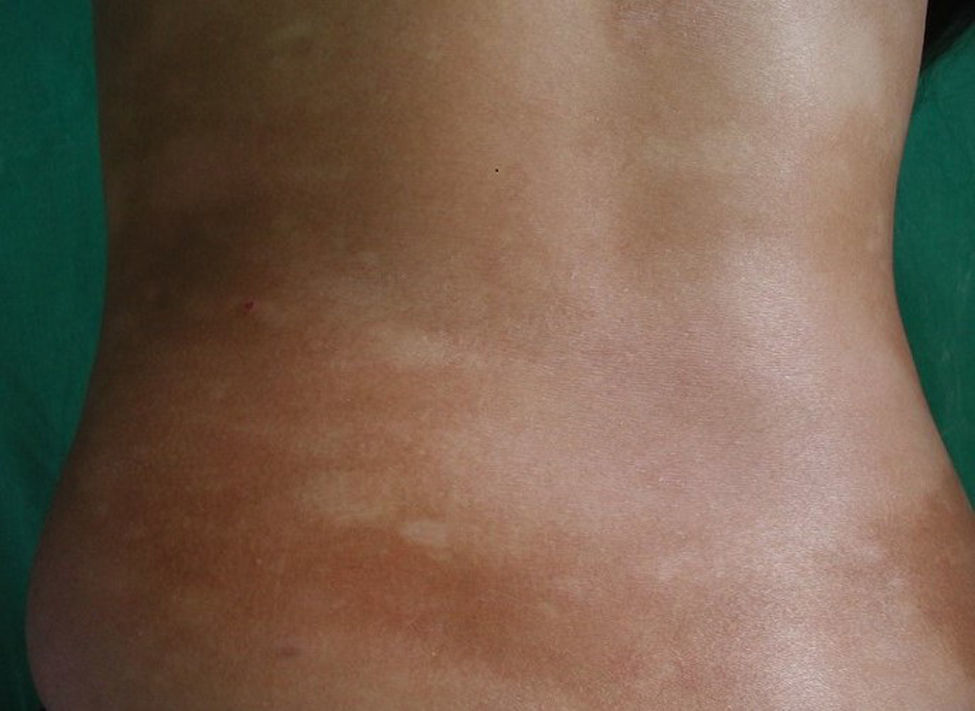
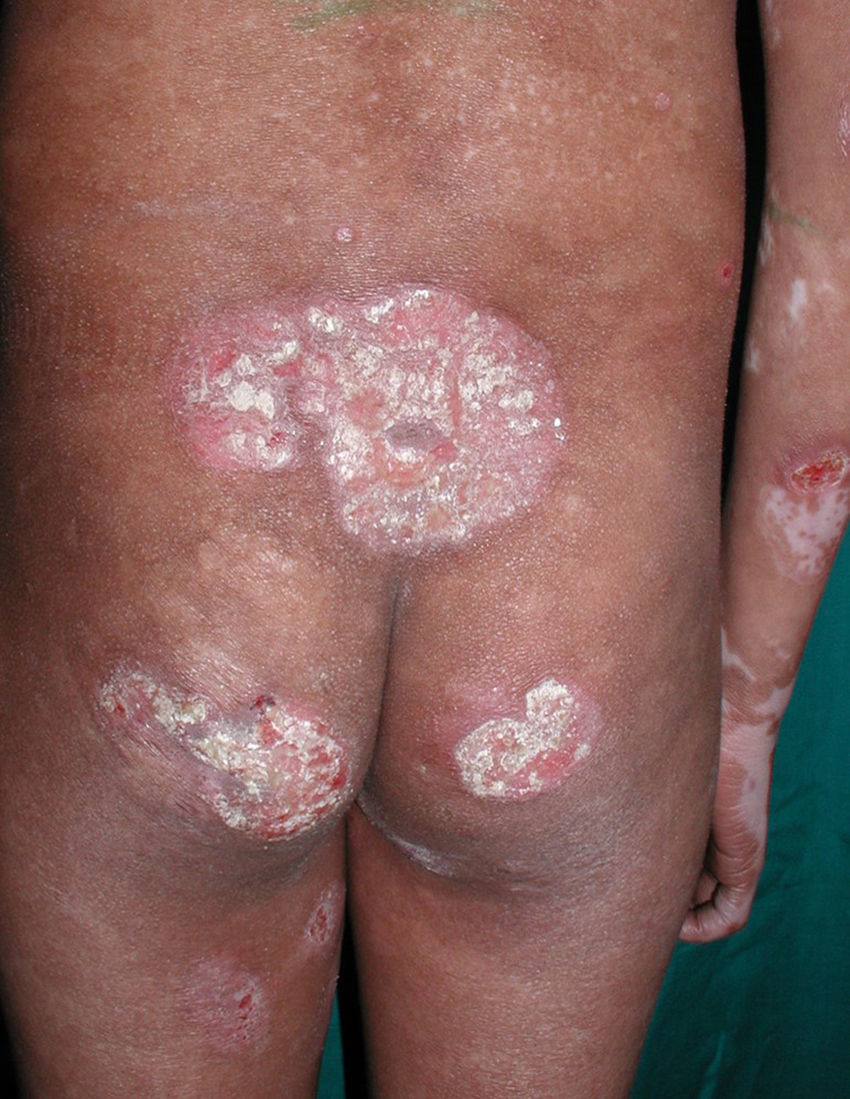
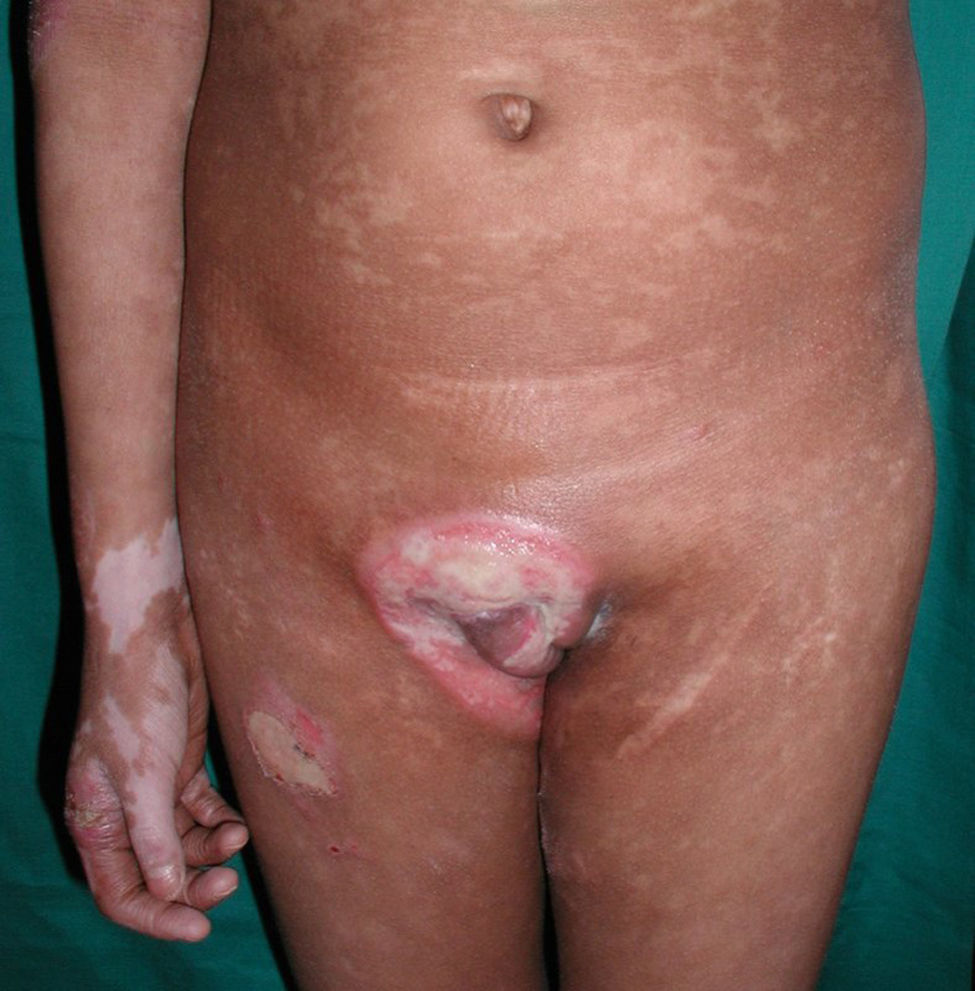
En relación con los antecedentes de la enfermedad se observó en 2 pacientes con diagnóstico clínico e histopatológico de PLC su evolución a MF después de 7 años de evolución (n.° 11 y n.° 12). Además, un paciente (n.° 5) con diagnóstico inicial de PL evolucionó a MF después de 5 años.
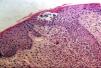
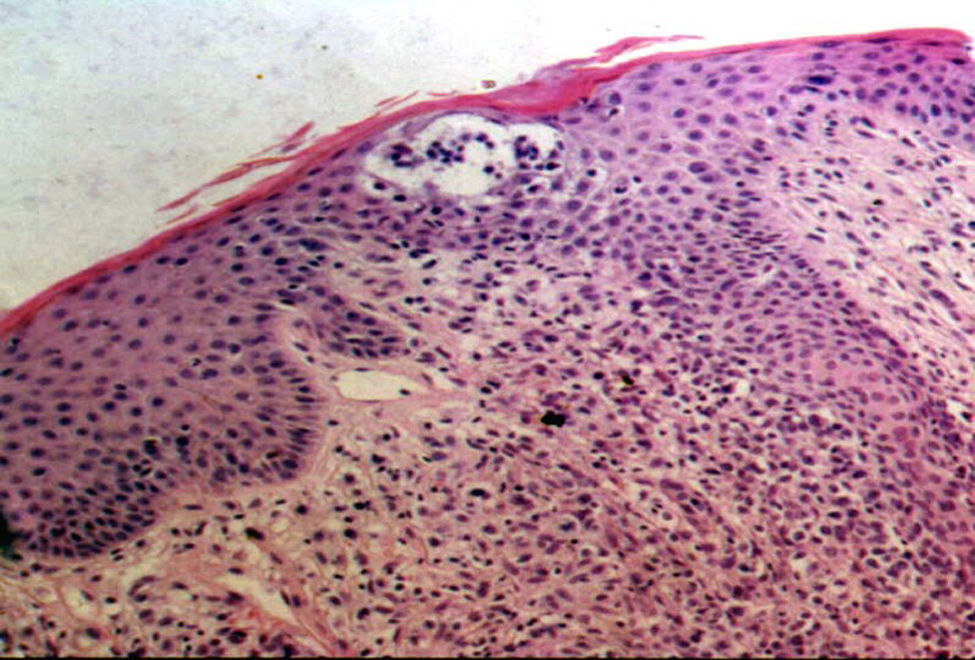
La totalidad de los pacientes presentó hallazgos histopatológicos compatibles con MF (infiltrado de linfocitos atípicos con epidermotropismo) y el 50% de los mismos presentó microabscesos de Pautrier (fig. 6). Uno de ellos (n.° 8) presentó una variante histopatológica foliculotrópica, sin correlación clínica de la misma. En cuanto a la inmunohistoquímica el 50% (7pacientes) presentó un inmunofenotipo de CD8+CD4–; 5 casos tuvieron CD8+ y CD4+y solo 2 fueron CD8– y CD4+(tabla 2).
En cuanto a los estudios para detectar reordenamientos clonales para el TCR, solo fueron realizados en 6 pacientes: n.° 1 (Vb22 CD4, región variable beta), n.° 4 (gamma), n.° 14 (beta) y en los números 3, 8 y 9 donde resultaron negativos. Todos fueron analizados en muestras de piel, excepto el caso n.° 4 que también fue estudiado en sangre y fue positivo para TCR delta (tabla 2).
El 78,5% de los pacientes (11p) se encontraba en el momento del diagnóstico en estadio IB, 2 pacientes en el IA y uno en estadio IVA2 (tabla 2). Este último presentó compromiso extracutáneo a nivel ganglionar axilar e inguinal.
En 11 de los 14 pacientes se pudo disponer de los valores de LDH y el recuento de eosinófilos. Ninguno de ellos presentó LDH elevada, y solo uno (n.° 4) tuvo hipereosinofilia (>700/mm3). Si bien este paciente tuvo episodios bronquiales crónicos, es llamativo que no presentó hipereosinofilia en su período libre de enfermedad.
En la tabla 2 se enumeran todos los tratamientos utilizados. Es importante hacer hincapié en que algunos de los esquemas terapéuticos se utilizaron de forma concurrente o sucesiva en los diferentes pacientes. El tratamiento de elección inicial en 12 (85,7%) pacientes fue la fototerapia, debido a la extensión de las lesiones: 6 recibieron PUVA, 2 UVB de banda estrecha (UVB-NB) y 3 ambas formas de fototerapia de forma consecutiva. Además, un paciente recibió fototerapia con UVA-1.
De forma concomitante y/o consecutiva, 9 pacientes también utilizaron como tratamiento: corticoides tópicos de alta potencia (clobetasol) (n=6), bexaroteno tópico (n=2) y tratamientos con retinoides orales (acitretín), psoralenos y helioterapia de forma combinada (n=1). El paciente en estadio IV recibió tratamiento de quimioterapia protocolo CHOP (ciclofosfamida 750mg/m2/día, doxorrubicina 50mg/m2/día, vincristina 1,4mg/m2/día IV y metilprednisona 60mg/m2/día por 5 días) y radioterapia electron beam.
Con respecto a la evolución, 8 pacientes se encuentran actualmente en tratamiento, 4 están libres de enfermedad, un paciente fue derivado a un hospital de adultos (n.° 1) después de haber estado libre de enfermedad durante 4 años y un paciente tuvo pérdida de seguimiento (n.° 2).
De los 14 pacientes 4 tuvieron por lo menos una recaída (n.° 1, 3, 4 y 5) y 3 presentaron progresión de su enfermedad a nivel cutáneo (n.° 4, 5 y 9) y uno (n.° 4) tuvo respuesta parcial con persistencia de la enfermedad. No hubo óbitos en nuestra serie de casos en una mediana de tiempo de seguimiento de 3 años (2 meses a 9 años), y la mediana de meses libre de enfermedad fue de 3 meses (uno a 48 meses). Es importante destacar que los amplios rangos se deben a que 9 de los 14 casos se han incorporado en la serie en los últimos 18 meses. De estos, 6 pacientes tienen menos de un año de seguimiento, encontrándose la mayoría de ellos en el primer tratamiento, por lo cual no podemos evaluar la efectividad terapéutica de forma global.
DiscusiónLa MF infantil representa entre el 5-16% del total de los pacientes con este diagnóstico, aunque de acuerdo a las publicaciones existen grandes variaciones en los porcentajes6–9,11,13. La incidencia antes de los 20 años sería aproximadamente de 0,05 por 100.000 habitantes por año14, siendo el LCP de células T de aparición más frecuente en niños. Si bien en la literatura se ha visto una predilección por el sexo masculino, en nuestra casuística tuvimos un leve predominio del género femenino13,14. Los hallazgos en el promedio de edad al diagnóstico (11,23 años) y el tiempo promedio de evolución hasta el momento del diagnóstico (3 años y 6 meses) son similares a lo que describen las otras series4,6–10,13,15.
El diagnóstico clínico de MF en la infancia es difícil de realizar, dado la baja frecuencia de aparición de esta entidad, como así también el bajo grado de sospecha por parte de los médicos y la multiplicidad de diagnósticos diferenciales que deben tenerse en cuenta. Esta situación se ha visto reflejada en nuestro estudio, en la demora para realizar el diagnóstico (3 años y 6 meses) y la baja sospecha clínica en la primera consulta (solo 57% de los mismos)4,9.
La variante clínica más frecuente en la infancia es la hipopigmentada, especialmente en pacientes con fototipo de piel III-IV4,5,7,8,10,11,13,16,17, lo cual es coincidente con la casuística presentada, coexistiendo con lesiones de MF clásica en el 40% de nuestros pacientes, como ya se ha descrito en otras publicaciones6,8,10. Es importante destacar que un paciente con clínica de PLC tuvo diagnóstico histopatológico de MF; y 2 pacientes con diagnóstico de PLC y uno de PL evolucionaron con los años a MF, situaciones que están descritas de forma infrecuente en la literatura médica.
La MF clásica habitualmente tiene un inmunofenotipo CD3, CD4, CD5+ y CD8–, con pérdida de expresión de CD7. Asimismo, en la variante hipopigmentada es frecuente observar la predominancia de CD8+, coincidiendo con lo hallado en nuestros pacientes (12p)5,7,9. Es de resaltar que el 35% de los casos (5p), presentó CD4 y CD8+, lo cual es una variante poco frecuente18,19.
La respuesta al tratamiento de nuestros pacientes es difícil de analizar debido a que es un trabajo retrospectivo y que la mayoría de los niños se encuentran aún realizando tratamiento. El 64% de los casos se han incorporado a la serie en los últimos 18 meses y casi la mitad de los pacientes tienen menos de un año de seguimiento.
Los tratamientos para MF varían según el estadio. Debido a que la mayoría de los pacientes presenta un compromiso cutáneo generalizado sin afectación sistémica, es de elección el uso de la fototerapia. En los casos localizados se emplean los corticoides tópicos. La fototerapia fue el tratamiento de elección en la mayoría de nuestros pacientes, siendo PUVA más eficaz que UVB-BE, sin efectos adversos detectados hasta la actualidad. Sin embargo, los pacientes en este grupo de edad requerirán a lo largo de su vida múltiples tratamientos, debido a la alta frecuencia de recidivas que presenta esta enfermedad en su evolución. Por dicha causa, en la actualidad, se aconseja realizar fototerapia de mantenimiento por períodos prolongados para evitar las recaídas tempranas5,13,14,17. Estos esquemas de tratamiento, si bien prolongan los períodos libres de enfermedad, puede llevar a dosis acumulativas altas de UVA, siendo importante la monitorización a largo plazo por la posibilidad de carcinogénesis a edades tempranas16. Más de la mitad de nuestros pacientes realizaron tratamientos locales con corticoides de alta potencia y/o bexaroteno de forma tópica, siendo la respuesta regular o poco sostenida en el tiempo.
Acorde con la literatura, todos los pacientes, excepto uno, presentaron estadio IA o IB2,4,6,8–11,13,15,16. El 78% de los pacientes presentaron estadio IB, cuyo pronóstico es excelente, con una expectativa de vida comparable a la de la población general1,20. La evolución en la mayoría de nuestros pacientes ha sido muy buena, sin óbitos y con progresión de la enfermedad únicamente en el 20% de los mismos. Esta progresión solo se manifestó con mayor extensión cutánea, pero sin compromiso sistémico. Un único paciente presentó tumores cutáneos y afectación nodal, encontrándose libre de enfermedad después de 7 años de seguimiento. Otras series de MF infantil muestran similares resultados, sin progresión de la enfermedad5,13,15.
Las limitaciones en nuestra casuística son las de un trabajo retrospectivo. Se han utilizado definiciones empíricas en cuanto a la respuesta al tratamiento, ya que no están estandarizadas en la infancia, por lo que dependen fundamentalmente de la valoración del profesional a cargo. También es limitante el corto período de seguimiento, lo cual hace difícil sacar conclusiones de la evolución a largo plazo, si bien existen publicaciones que demuestran poca progresión de la enfermedad y una excelente supervivencia5,9,11,13,14. Sin embargo, en el trabajo de Ai et al.14 en pacientes menores de 30 años, con un seguimiento a 10 años, se evidenció un riesgo aumentado de aparición de segundo cáncer (Standard Incidence Ratio 3,49), especialmente linfoma y melanoma, aunque la supervivencia global fue del 88,9% al 94,3% y las diferencias no fueron estadísticamente significativas.
En conclusión, la MF en niños es una entidad infrecuente y de difícil diagnóstico debido a la multiplicidad de diagnósticos diferenciales a tener en cuenta, siendo la variante hipopigmentada la forma de presentación más frecuente en este grupo de edad. La MF en la niñez, a pesar de tener un buen pronóstico, presenta altas tasas de recidiva y requiere, por lo tanto, un control a largo plazo. La monitorización en el tiempo de los pacientes pediátricos con MF ayudará a determinar la evolución y progresión de la misma, los efectos adversos a largo plazo de los tratamientos, así como la aparición de segundas neoplasias.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecimientos
Dr. Guillermo Chantada y Centro Médico PSORIAHUE.


