











Los linfomas cutáneos primarios son un grupo heterogéneo de procesos linfoproliferativos malignos que se manifiestan inicialmente en la piel sin evidencia de afectación extracutánea en el momento del diagnóstico y que presentan una baja incidencia (7-10 casos×106 personas/año). Se dividen en linfomas cutáneos derivados de linfocitos T (70-85%) y de células B (15-30%). El reconocimiento de la idiosincrasia de los linfomas cutáneos primarios por parte de hematólogos y oncólogos es cada vez mayor, como queda reflejado en la última actualización de la clasificación de la Organización Mundial de la Salud, si bien todavía quedan matices o peculiaridades a considerar en su manejo que obligan a los dermatólogos a seguir trabajando para una plena integración de las diferentes situaciones clínicas que nos plantean en futuras revisiones de la clasificación de las neoplasias linfoides. El diagnóstico de un linfoma cutáneo primario se establece en base a los hallazgos clínicos, histopatológicos, inmunofenotípicos y genotípicos (demostración de monoclonalidad linfoide T o B) de las lesiones cutáneas y en el resultado de las distintas exploraciones complementarias destinadas a descartar una afectación extracutánea.
CD30+ primary cutaneous lymphomas comprise a large group of malignant lymphoproliferative disorders that present in the skin without extracutaneous involvement at the time of diagnosis. The incidence of these lymphomas is low, at 7 to 10 cases per 100 000 population. Two types, derived from T cells (70%–85%) or B cells (15%–30%), have been identified. Hematologists and oncologists have increasingly recognized the idiosyncrasy of primary cutaneous lymphomas, as reflected in the updated classification of the World Health Organization. However, there remain nuances or small differences to consider when managing these conditions, obliging dermatologists to continue to strive to fully reconcile the various clinical pictures in future reviews of the classification of lymphoid neoplasms. A diagnosis of a primary cutaneous lymphoma is based on clinical, histopathologic, immunophenotypic, and genotypic criteria, particularly evidence of T- or B-cell lymphoid monoclonality in lesions. Also relevant are complementary tests to rule out extracutaneous involvement.
Los linfomas cutáneos primarios (LCP) son un grupo heterogéneo de procesos linfoproliferativos malignos que se manifiestan inicialmente en la piel sin evidencia de afectación extracutánea en el momento del diagnóstico. Presentan una baja incidencia (7-10 casos×106/año)1,2 y se dividen en LCP derivados de linfocitos T (LCCT) (70-85%) y de células B (LCCB) (15-30%)3–5.
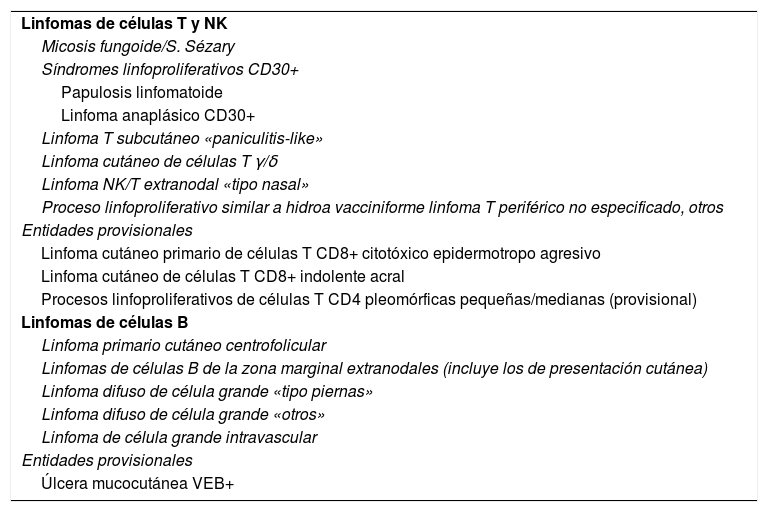
El reconocimiento de la idiosincrasia de los LCP por parte de hematólogos y oncólogos es cada vez mayor, como queda reflejado en la última actualización de la clasificación de la Organización Mundial de la Salud (OMS) (tabla 1)6. Sin embargo, quedan todavía matices o peculiaridades a considerar en el manejo de los LCP que obligan a los dermatólogos a seguir trabajando para conseguir una plena integración de las diferentes situaciones clínicas en las futuras revisiones de la clasificación de la OMS de las neoplasias linfoides (clasificación consensuada OMS/EORTC de los linfomas cutáneos 2018)1. Los cambios más relevantes de la clasificación OMS 2016 incluyen: a) El LCCT CD4+ pleomórfico de células de pequeño/mediano, la clasificación actual lo define como «proceso linfoproliferativo cutáneo primario de células pleomórficas de pequeño/mediano», perdiendo su condición de «linfoma» verdadero; b) Se definen algunas nuevas entidades provisionales como el LCCT de células CD8+ acral o la úlcera mucocutánea por virus de Epstein-Barr (VEB), mientras que otras entidades como el LCCT CD8+ epidermotropo agresivo mantienen su carácter provisional. Respecto a los LCCB, la última actualización de la clasificación OMS defiende la idiosincrasia del LCCB de células del centro-folicular y del LCCB difuso de células grandes «tipo piernas» como entidades diferenciadas, pero no consideran el LCCB cutáneo primario de la zona marginal como una entidad individualizada y lo incluyen dentro de los linfomas extranodales MALT (tabla 1).
Clasificación OMS 2016: subtipos de linfomas cutáneos primarios reconocidos
| Linfomas de células T y NK |
| Micosis fungoide/S. Sézary |
| Síndromes linfoproliferativos CD30+ |
| Papulosis linfomatoide |
| Linfoma anaplásico CD30+ |
| Linfoma T subcutáneo «paniculitis-like» |
| Linfoma cutáneo de células T γ/δ |
| Linfoma NK/T extranodal «tipo nasal» |
| Proceso linfoproliferativo similar a hidroa vacciniforme linfoma T periférico no especificado, otros |
| Entidades provisionales |
| Linfoma cutáneo primario de células T CD8+ citotóxico epidermotropo agresivo |
| Linfoma cutáneo de células T CD8+ indolente acral |
| Procesos linfoproliferativos de células T CD4 pleomórficas pequeñas/medianas (provisional) |
| Linfomas de células B |
| Linfoma primario cutáneo centrofolicular |
| Linfomas de células B de la zona marginal extranodales (incluye los de presentación cutánea) |
| Linfoma difuso de célula grande «tipo piernas» |
| Linfoma difuso de célula grande «otros» |
| Linfoma de célula grande intravascular |
| Entidades provisionales |
| Úlcera mucocutánea VEB+ |
Fuente: Willemze et al.1
El diagnóstico de LCP suele establecerse en base a los hallazgos clínicos, histopatológicos, inmunofenotípicos y genotípicos (demostración de monoclonalidad linfoide T o B) de las lesiones cutáneas y en el resultado de las distintas exploraciones complementarias destinadas a descartar una afectación extracutánea5,7,8. Las exploraciones complementarias para la evaluación inicial de los LCP se resumen en las tablas 2 y 3 y la estadificación en las tablas 4 y 5.
Exploraciones complementarias en el diagnóstico y evaluación de la micosis fungoide/síndrome de Sézary
| Estadios IA-IB-IIA |
| Hemograma, morfología sangre periférica |
| Bioquímica estándar (LDH) |
| Serología (HIV, HTLV-I) |
| Inmunofenotipo linfocitos T sangre periférica (ratio CD4/CD8) y estudio clonalidad TCR |
| Pruebas epicutáneas de contacto |
| Radiografía de tórax |
| Ecografía ganglionar |
| Biopsia ganglionara |
| Estadios IIB-III-IV |
| TAC±PET |
| Inmunofenotipo linfocitos T sangre periférica (ratio CD4/CD8) y estudio clonalidad TCR |
| Biopsia ganglionara |
| Considerar biopsia de médula ósea |
Biopsia de adenopatías en los siguientes escenarios clínicos: tamaño mayor de 1,5cm; consistencia dura o irregularidades morfológicas en ecografía; agrupados o adheridos; elevada actividad funcional por PET.Fuente: Olsen et al.7.
Exploraciones complementarias para el diagnóstico y evaluación de los linfomas cutáneos de células T distintos de micosis fungoide/síndrome de Sézary
| Historia clínica completa/revisión por sistemas y exploración física |
| Estudios de laboratorio |
| Hemograma completo, bioquímica estándar, LDH sérica |
| Citometría de flujo de células mononucleares en sangre periférica y estudio clonalidad T |
| Estudios de imagena |
| TAC toraco-abdominal-pélvico con contraste o PET; incluyendo TAC o ecografía cervical si clínicamente indicada |
| Aspirado/biopsia de médula óseab |
| Requerido en linfomas cutáneos de comportamiento intermedio a agresivo |
| Podría considerarse en linfomas cutáneos con comportamiento clínico indolente, pero no requerido a no ser que estuviera indicado a partir de otras valoraciones de estadificación |
| Estudios adicionales según indicación clínica |
Ganglios linfáticos >1,0cm en su eje corto y/o PET con un aumento significativo de actividad deberían ser biopsiados (preferentemente biopsia escisional).
En el momento de esta propuesta, no existe un estándar unificado para el estudio de médula ósea como parte de la valoración de estadificación en los linfomas cutáneos de evolución clínica indolente. Los clínicos deberían seguir las guías prácticas de tratamiento convencionales de cada país.Fuente: Kim et al.8.
Estadificación TNM(B) de la micosis fungoide/síndrome de Sézary
| Afectación cutánea (T) | |
| T1 | Placas, pápulas o placas limitadas a <10% de la superficie corporal (T1a=máculas; T1b=placas±máculas) |
| T2 | Lesiones en ≥10% de la superficie corporal (T2a=máculas; T2b=placas±máculas) |
| T3 | Uno o más tumores (≥1cm) |
| T4 | Eritrodermia ≥80% de la superficie corporal |
| Afectación nodal (N) | |
| N0 | No adenopatías clínicas |
| N1 | Adenopatías clínicas. Grado 1, NCI LN O-2 |
| N1a | No clonal |
| N1b | Clonal |
| N2 | Adenopatías clínicas. Grado 2, NCI LN 3 |
| N2a | No clonal |
| N2b | Clonal |
| N3 | Adenopatías clínicas. Grado 3-4, NCI LN 4, clonal o no |
| Nx | Adenopatías clínicas. No confirmación histopatológica |
| Afectación visceral (M) | |
| M0 | No afectación visceral |
| M1 | Afectación visceral con confirmación histológica |
| Sangre periférica (B) | |
| B0 | No afectación significativa de sangre periférica: ≤5% células de Sézary, <250/μL células de Sézary, CD4+/CD26− o CD4+/CD7− o CD4+/CD26− y CD4+/CD7− en <15% células por citometría |
| B0a | No clona |
| B0b | Clona |
| B1 | Baja carga tumoral en sangre periférica (no cumple criterios de B0 ni B2) |
| B1a | No clona |
| B1b | Clona |
| B2 | Alta carga tumoral en sangre periférica: Clona + uno de los siguientes criterios: >1.000 células de Sézary/μL, ratio CD4/CD8≥10, CD4+/CD7− ≥40% o CD4+/CD26− ≥30% |
| T | N | M | B | |
|---|---|---|---|---|
| IA | 1 | 0 | 0 | 0,1 |
| IB | 2 | 0 | 0 | 0,1 |
| II | 1,2 | 1,2 | 0 | 0,1 |
| IIB | 3 | 0-2 | 0 | 0,1 |
| III | 4 | 0-2 | 0 | 0,1 |
| IIIA | 4 | 0-2 | 0 | 0 |
| IIIB | 4 | 0-2 | 0 | 1 |
| IVA1 | 1-4 | 0-2 | 0 | 2 |
| IVA2 | 1-4 | 3 | 0 | 0-2 |
| IVB | 1-4 | 0-3 | 1 | 0-2 |
Fuente: Olsen et al.7.
Estadificación TNM de los linfomas cutáneos primarios de células T distintos a la micosis fungoide/síndrome de Sézary
| Afectación cutánea (T) |
| T1: Lesión cutánea solitaria |
| T1a: <5cm |
| T1b: >5cm |
| T2: Lesiones múltiples agrupadas en una sola región anatómica o en 2 contiguas |
| T2a: <15cm diámetro total del área afectada |
| T2b: >15 y <30cm diámetro total del área afectada |
| T2c: >30cm diámetro total del área afectada |
| T3: Lesiones múltiples generalizadas |
| T3a: Lesiones en >2 zonas anatómicas no contiguas |
| T3b: Lesiones en ≥3 regiones anatómicas |
| Afectación nodal (N) |
| No: No afectación nodal clínica o patológica |
| N1: Afectación nodal periférica de 1 ganglio en territorio de drenaje linfático de la lesión cutánea |
| N2: Afectación de 2 ganglios o 1 ganglio periférico en zona no de drenaje linfático de la lesión cutánea |
| N3: Afectación de ganglio(s) linfático central |
| Afectación visceral (M) |
| M0: No afectación visceral |
| M1: Afectación visceral |
Fuente: Kim et al.8.
Dentro del grupo de los LCCT se incluyen la micosis fungoide (MF), el subtipo más frecuente representando aproximadamente la mitad de los mismos, el síndrome de Sézary (SS) y los LCCT distintos al grupo MF/SS, grupo heterogéneo de entidades caracterizadas por una proliferación maligna de células T3,4.
Micosis fungoideLa MF es el subtipo más frecuente de LCP con una incidencia anual de 0,3-0,5 casos nuevos por 100.000 habitantes cada año. Afecta a pacientes adultos con una predilección (2:1) por el sexo masculino. La MF es una proliferación monoclonal de linfocitos T cooperadores maduros de memoria CD4+/CD45RO+2.
Manifestaciones clínicasEn sus estadios iniciales, la MF permanece localizada en la piel durante años en forma de máculas eritemato-descamativas o placas infiltradas persistentes, habitualmente localizadas en áreas no expuestas. En estas fases iniciales (estadios IA, IB, IIA) la enfermedad no ocasiona un impacto en la supervivencia. En un porcentaje limitado de pacientes (20%) la enfermedad progresa a estadios más avanzados desarrollando nódulos o tumores, solitarios o múltiples, con tendencia a la ulceración (estadio IIB), que suelen coexistir simultáneamente con máculas o placas y/o afectación ganglionar y visceral específica (estadio IV) (fig. 1A-C). Se consideran datos con valor pronóstico clínico negativo en la MF una edad superior a 60 años, unos niveles elevados de LDH, la presencia de afectación visceral, o de transformación histológica a un linfoma de células grandes9–11.
 Lesión de micosis fungoide (MF) en fase macular. (B) Lesión de MF tipo placa. (C) MF tumoral. (D) Histología de MF en placa con infiltrado en banda de dermis reticular superficial y media con linfocitos atípicos de gran tamaño. Asocia fenómenos de epidermotropismo. (E) El indicador de proliferación Ki67 permite detectar las células linfoides de gran tamaño. (F) Infiltrado difuso en toda la dermis correspondiente a una lesión de MF tumoral. (G) Células de gran tamaño y núcleo pleomórfico de una biopsia de MF tumoral. (H) Índice de proliferación >50% en una lesión de MF tumoral.")
(A) Lesión de micosis fungoide (MF) en fase macular. (B) Lesión de MF tipo placa. (C) MF tumoral. (D) Histología de MF en placa con infiltrado en banda de dermis reticular superficial y media con linfocitos atípicos de gran tamaño. Asocia fenómenos de epidermotropismo. (E) El indicador de proliferación Ki67 permite detectar las células linfoides de gran tamaño. (F) Infiltrado difuso en toda la dermis correspondiente a una lesión de MF tumoral. (G) Células de gran tamaño y núcleo pleomórfico de una biopsia de MF tumoral. (H) Índice de proliferación >50% en una lesión de MF tumoral.
El estudio histopatológico de las máculas iniciales muestra un infiltrado en la dermis superficial formado por linfocitos pequeños cerebriformes que con frecuencia se distribuyen siguiendo la capa basal epidérmica (epidermotropismo) sin cambios espongióticos. La progresión de las lesiones a placas da lugar a un infiltrado más denso en banda en la dermis papilar, y a un epidermotropismo más intenso (microabscesos de Pautrier intraepidérmicos). Las lesiones tumorales se caracterizan por densos infiltrados monomorfos de linfocitos atípicos, nodulares o difusos, afectando todo el grosor de la dermis. La presencia de células grandes (habitualmente CD30+) es frecuente y cuando estas representan más del 25% del infiltrado, se considera que existe transformación a célula grande (fig. 1D-H). El fenotipo habitual es CD2+, CD3+, βF1+, CD4+, CD8–, CD45RO con una pérdida de marcadores de linfocitos T maduros (CD7, CD2 o CD5). Un fenotipo citotóxico (CD8+, TIA1+ o γδ+) en la MF no suele implicar diferencias clínicas, pronósticas o evolutivas. El estudio de clonalidad mediante técnicas de PCR detecta un reordenamiento monoclonal del receptor de la célula T (TCR) en la mayoría de los casos3.
Mecanismos patogénicosLos pacientes con MF/SS presentan alteraciones genómicas complejas, heterogéneas, poco específicas, que afectan a genes implicados en la activación de la célula T, apoptosis, remodelación de la cromatina o de respuesta al daño del DNA, así como genes reguladores de ciclo celular (TP53, PLCG1, CARD11, STAT5B, NFKB2, IL6, ITPR1, RASA2, TNFRSF10A, FASN, ZEB1, DNMT3A o KMT2C). De forma concreta o específica parece clave la activación de vías de señalización como STAT, NOTCH1 o β-catenina (vía NF-kB)12–20.
Variantes de micosis fungoideMicosis fungoide foliculotropaLa MF foliculotropa (MFF) es una variante clínico-patológica peculiar caracterizada por un tropismo preferente por el epitelio del folículo pilosebáceo. Puede asociarse con mucinosis folicular. Las lesiones suelen localizarse en la cara, cuello y tronco, y se manifiestan como pápulas foliculares agrupadas, quistes, comedones, áreas alopécicas y/o lesiones seudotumorales (fig. 2A y B). La supervivencia global a los 10 años es de aproximadamente el 82% frente al 91% de la forma clásica, y del 41% frente a un 90% a los 15 años. Sin embargo, recientemente se ha propuesto la existencia de distintos subgrupos de MFF: una forma precoz o «superficial» con componente neoplásico poco prominente y con un pronóstico similar a la MF clásica en sus estadios iniciales, y una variante «profunda» de evolución más agresiva21,22. Se conoce como MF siringotropa a aquellas formas de MF con una afectación exclusiva o predominante de las glándulas sudoríparas ecrinas. Las lesiones suelen consistir en placas infiltradas, hiperpigmentadas y anhidróticas y, a menudo, alopécicas. La MF siringotropa puede desarrollarse bien de forma aislada o bien asociada a una MFF (MF anexotropa).
Reticulosis pagetoide Lesiones papulares y comedogénicas de una micosis fungoide foliculotropa (MFF). (B) Histología de una lesión papular de MFF incipiente o superficial que muestra foliculotropismo linfoide y fenómenos de mucinosis folicular.")
La reticulosis pagetoide (RP) es una variante localizada de MF, caracterizada clínicamente por una o varias placas hiperqueratósicas generalmente en áreas distales de las extremidades, e histopatológicamente por un intenso epidermotropismo. Las células neoplásicas presentan un fenotipo CD8+ o CD4+ y con frecuencia son CD30+. Debe diferenciarse de otros subtipos de LCCT agresivos con epidermotropismo prominente, ya que la RP es un proceso de evolución clínica indolente, proponiéndose la radioterapia localizada como el tratamiento de elección1.
Piel laxa granulomatosaLa piel laxa granulomatosa o granulomatous slack skin disease es una variante peculiar y excepcional de MF caracterizada clínicamente por el desarrollo lento y progresivo de piel redundante en los grandes pliegues (axilas, ingles). Histológicamente, se observa un denso infiltrado de características granulomatosas con densos infiltrados linfoides dérmicos y abundantes células gigantes multinucleadas con fenómenos de elastofagocitosis y elastólisis. Suele observarse asimismo un infiltrado en banda superficial con células linfoides atípicas y un grado variable de epidermotropismo1.
Síndrome de SézaryEl SS es un proceso clásicamente caracterizado por una eritrodermia, linfadenopatías generalizadas y más de 1.000/mm3 (o >10%) células mononucleares atípicas circulantes con núcleo cerebriforme (células de Sézary). Se observa casi exclusivamente en adultos de edad avanzada y presenta una cierta predisposición por el sexo masculino. Junto a una eritrodermia difusa (eritema que afecta a >80% de la superficie corporal) asociada a un intenso prurito, suele acompañarse de ectropión, queratodermia palmo-plantar, distrofia ungueal y alopecia (fig. 3). Las células de Sézary circulantes son CD4+, CD7−, CD26− con una relación entre linfocitos CD4+ y CD8+ >10. La evidencia de una expansión clonal CD4+/CD7− ≥40%, o CD4+/CD26− ≥30%, constituiría también un criterio suficiente para el diagnóstico. Los hallazgos histopatológicos cutáneos del SS son similares a los observados en la MF, aunque suelen presentar un epidermotropismo de menor intensidad. La enfermedad suele seguir una evolución agresiva con una supervivencia a los 5 años que no supera el 30-40%7,10,23.
Tratamiento de la micosis fungoide y del síndrome de Sézary
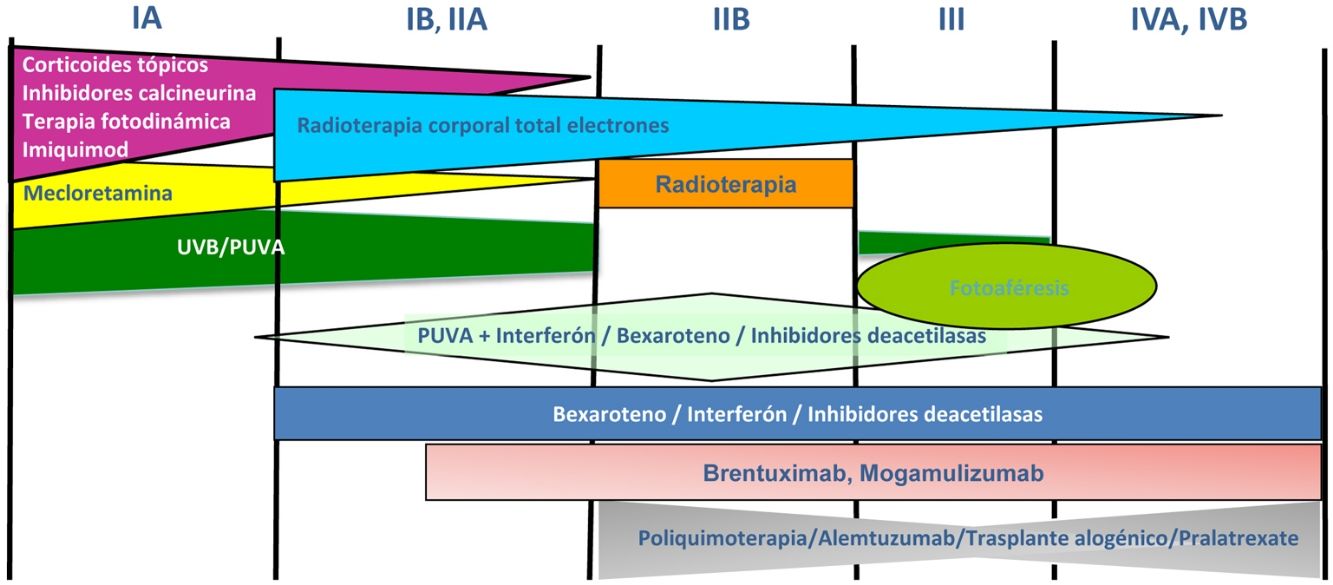
Los pacientes con estadios IA-IIA deben ser tratados inicialmente con terapias dirigidas a la piel, como los corticoides tópicos o la fototerapia (PUVA-terapia o UVB de banda estrecha)24,25. Los citostáticos tópicos (mecloretamina o carmustina) permiten obtener respuestas terapéuticas superiores a un 50% de los casos, aunque su uso está limitado por su mala tolerancia (dermatitis irritativa)26. Otros tratamientos tópicos incluyen el bexaroteno en gel (no financiado), el resiquimod (agonista de receptores Toll-like, no aprobado) y ocasionalmente inhibidores de la calcineurina (no aprobados) o la terapia fotodinámica27,28. En aquellos pacientes con placas infiltradas diseminadas, o en los casos refractarios a los tratamientos anteriores, suele prescribirse bexaroteno oral29 o interferón-α30 en monoterapia, o bien, tratamientos combinados con bexaroteno, interferón-α o PUVA-terapia24. La irradiación cutánea total con electrones representa una alternativa terapéutica eficaz en casos seleccionados de MF estadios IB o IIA31–33 (fig. 4). En la MFF, los mejores resultados parecen obtenerse con el empleo de PUVA asociado a bexaroteno o interferón-α o mediante irradiación cutánea total con electrones34.

En pacientes con tumores únicos o localizados (IIB) se emplea radioterapia localizada y en caso de lesiones múltiples, gemcitabina o doxorrubicina en monoterapia3,35,36. Otros tratamientos modificadores de la respuesta biológica como los inhibidores de la deacetilasa de las histonas (vorinostat, romidepsina) utilizados tanto para enfermedad avanzada como inicial, en monoterapia o en combinación con otros tratamientos, pueden ser de utilidad, si bien aún no están aprobados en la Unión Europea37. Las pautas de poliquimioterapia (CHOP) solo están indicadas en los pacientes con diseminación ganglionar y/o visceral (estadio IV) refractarios a las terapias anteriores o dentro del contexto de un tratamiento previo al trasplante alogénico de precursores hematopoyéticos. Recientemente han sido aprobados brentuximab vedotin (anticuerpo monoclonal dirigido contra el antígeno CD30) y mogamulizumab (anticuerpo monoclonal contra el receptor CCR4) en pacientes con MF avanzada y SS38,39 (fig. 4).
La fotoféresis extracorpórea se considera el tratamiento inicial del SS tras el diagnóstico40. Puede asociarse a interferón-α, bexaroteno, irradiación corporal total o PUVA-terapia. Metotrexato, prednisona y clorambucilo son terapias clásicas del SS, aunque poseen una eficacia relativa. En pacientes refractarios, el manejo de SS avanzado no difiere significativamente del de una MF avanzada, si bien algunos fármacos como alemtuzumab (anticuerpo monoclonal contra el antígeno CD52) y mogamulizumab podrían tener un perfil de eficacia/toxicidad más favorable para casos refractarios de SS41, habiendo este último demostrado una mejor respuesta terapéutica en el compartimento sanguíneo (a diferencia de brentuximab, que tendría una mayor eficacia sobre el compartimento cutáneo)23,24,38,39 (fig. 4).
Finalmente, en pacientes con MF/SS en fases avanzadas, debería considerarse la posibilidad de un trasplante alogénico de precursores hematopoyéticos42 o su posible inclusión en ensayos clínicos, dado que en el momento actual existen diversas moléculas en desarrollo clínico dirigidas tanto a controlar la respuesta inmune (anticuerpos monoclonales anti-PD1 o anti-PDL1) como a potenciar la citotoxicidad mediada por anticuerpos monoclonales (KIRDL2)43.
Proliferaciones linfoides cutáneas CD30 positivasLas proliferaciones linfoides cutáneas CD30+ son un grupo de procesos linfoproliferativos cutáneos que poseen en común la expresión por parte de las células neoplásicas del antígeno de activación linfoide CD30. Este espectro de entidades incluye la papulosis linfomatoide (PL) y el linfoma de células grandes anaplásicas CD30+ (LCCT-CD30+). Suelen presentar un buen pronóstico con una supervivencia media superior al 90% a los 5 años1.
Papulosis linfomatoideManifestaciones clínicasSe caracteriza por la aparición recurrente y crónica de pápulas o nódulos que presentan una involución espontánea dejando frecuentemente cicatrices residuales. Las lesiones suelen ser pápulas eritematosas, con tendencia a presentar ulceración y necrosis central, de tamaño, número y distribución variables (lesiones aisladas, agrupadas o diseminadas) habitualmente localizadas en tronco y extremidades (fig. 5A). Entre el 5 y el 20% de pacientes pueden presentar una asociación con otro proceso neoplásico, generalmente hematológico (linfoma de Hodgkin) o MF44.
 Pápulas en diferente estadio evolutivo correspondientes a una papulosis linfomatoide (PL). (B) Infiltrado dérmico formado por linfocitos atípicos y rico en eosinófilos de una lesión papular de PL. (C) Linfocitos de gran tamaño y pleomorfismo nuclear de una lesión de PL. (D) Expresión del antígeno CD30 en los linfocitos de aspecto activado y gran tamaño en la PL.")
(A) Pápulas en diferente estadio evolutivo correspondientes a una papulosis linfomatoide (PL). (B) Infiltrado dérmico formado por linfocitos atípicos y rico en eosinófilos de una lesión papular de PL. (C) Linfocitos de gran tamaño y pleomorfismo nuclear de una lesión de PL. (D) Expresión del antígeno CD30 en los linfocitos de aspecto activado y gran tamaño en la PL.
La PL muestra un infiltrado linfoide de disposición perivascular e interfase con una disposición o morfología en «cuña» en la dermis, con ocasional extensión a tejido celular subcutáneo. En dicho infiltrado se observan células linfoides atípicas, de gran tamaño, que expresan el antígeno CD30 similares a las células de Reed-Sternberg del linfoma de Hodgkin. Se observa asimismo un denso infiltrado inflamatorio acompañante con abundantes polimorfonucleares neutrófilos y eosinófilos. Los hallazgos histopatológicos observados pueden plantear el diagnóstico diferencial con una pitiriasis liquenoide o incluso con picaduras de artrópodo. Las células neoplásicas expresan un fenotipo T cooperador (CD3+, CD4+, CD8−) con presencia de grupos de células atípicas de gran tamaño CD30+44 (fig. 5B-D).
En los últimos años se han descrito distintas variantes histopatológicas. Junto a las formas clásicas tipo A (histiocítica), tipo B (similar a MF) o tipo C (similar a LCCT-CD30+)44, se han descrito variantes con fenotipo citotóxico (tipo D)45 que pueden simular un linfoma cutáneo T citotóxico epidermotropo agresivo; variantes con histología angioinvasiva (tipo E)46 que suelen manifestarse clínicamente con lesiones ulcero-necróticas; formas predominantemente foliculotropas, granulomatosas, etc. Estas variantes no conllevan una distinta evolución clínica, aunque pueden plantear dificultades diagnósticas en su diferenciación con otros procesos linfoproliferativos. No se han descrito alteraciones genéticas específicas en la PL, aunque recientemente se han detectado reordenamientos que implican al gen DUSP22 con IRF4 en el locus 6p25.3 en un reducido número de casos47.
Valoración y tratamientoEl diagnóstico de la PL se establece en base a las características clínico-patológicas de las lesiones cutáneas y a su carácter autoinvolutivo. El tratamiento de la PL vendrá condicionado por la extensión y las características de las lesiones. En muchas ocasiones no se precisa tratamiento. En casos de lesiones extensas, necróticas o con tendencia a dejar cicatrices puede prescribirse tratamiento con metotrexato oral a dosis bajas (7,5 a 15mg/semana e incluso inferiores), fototerapia (UVB o PUVA-terapia) o interferón-α. Excepcionalmente, podría plantearse el tratamiento con brentuximab38,44.
Linfoma primario cutáneo anaplásico de células T CD30 positivasManifestaciones clínicasEl LCCT-CD30+ se presenta de forma característica como un nódulo solitario o múltiples nódulos de tamaño variable, agrupados o en distintas localizaciones anatómicas, de crecimiento rápido y con tendencia a la ulceración (fig. 6A). En su estadificación se aplica la clasificación TNM para los LCP distintos del grupo MF/SS (tabla 5). Aproximadamente un 10% de los pacientes pueden desarrollar afectación extracutánea, generalmente ganglionar locorregional44.
Histopatología Linfoma cutáneo primario anaplásico de células T CD30 positivas. (B) Infiltrado dérmico difuso de células linfoides atípicas. (C) Células linfoides atípicas y núcleos pleomórficos. (D) Expresión del antígeno CD30 en la mayor parte de las células del infiltrado.")
Histológicamente se caracteriza por una infiltración dérmica difusa formada por células linfoides atípicas de gran tamaño con abundante citoplasma, núcleos redondeados-ovales, y nucléolo prominente. Estas células presentan un fenotipo T CD4+/CD45RO+ con intensa expresión del antígeno CD30 (>75% de las células neoplásicas) (fig. 6B-D). La expresión de CD56 o marcadores citotóxicos no es excepcional (TIA-1, granzyme B) sin que ello tenga implicaciones pronósticas44.
En los LCCT-CD30+ no se detecta la translocación cromosómica t(2;5)(p23;q35), que caracteriza a los linfomas anaplásicos de origen nodal, ni la expresión de su proteína de fusión NPM/ALK (Anaplastic Lymphoma Kinase) asociada a dicha translocación. Tampoco expresan el antígeno EMA y son CD15– a diferencia de los linfomas anaplásicos ganglionares y del linfoma de Hodgkin. En aproximadamente un 25% de los casos, puede detectarse la translocación DUSP22/IRF444.
Pronóstico y tratamientoSuele ser un proceso de buen pronóstico incluso en aquellos casos con afectación ganglionar locorregional secundaria, con unas tasas de supervivencia a los 5 años superiores al 75-90%. Suele aconsejarse un tratamiento conservador mediante cirugía o radioterapia en lesiones solitarias, o bien metotrexato, interferón o monoquimioterapia en aquellos casos con lesiones localizadas. Una afectación cutánea multicéntrica extensa debe plantear la posibilidad de tratamiento con brentuximab y solo excepcionalmente otras alternativas más agresivas (poliquimioterapia)44.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


