



Presentamos el caso de un paciente de 40 años, sin antecedentes personales de interés, que consultó por un tumor periungueal en el segundo dedo de la mano izquierda. La lesión tenía un curso asintomático de dos años de evolución y un crecimiento lento y progresivo. La exploración mostraba un tumor de 1cm de diámetro, de bordes mal definidos y no adherido a planos profundos (fig. 1A). A la palpación la lesión era dura y levemente dolorosa. El paciente había presentado 4 años antes una lesión en la misma localización, de similares características clínicas, que fue extirpada con el diagnóstico de fibroma periungueal.
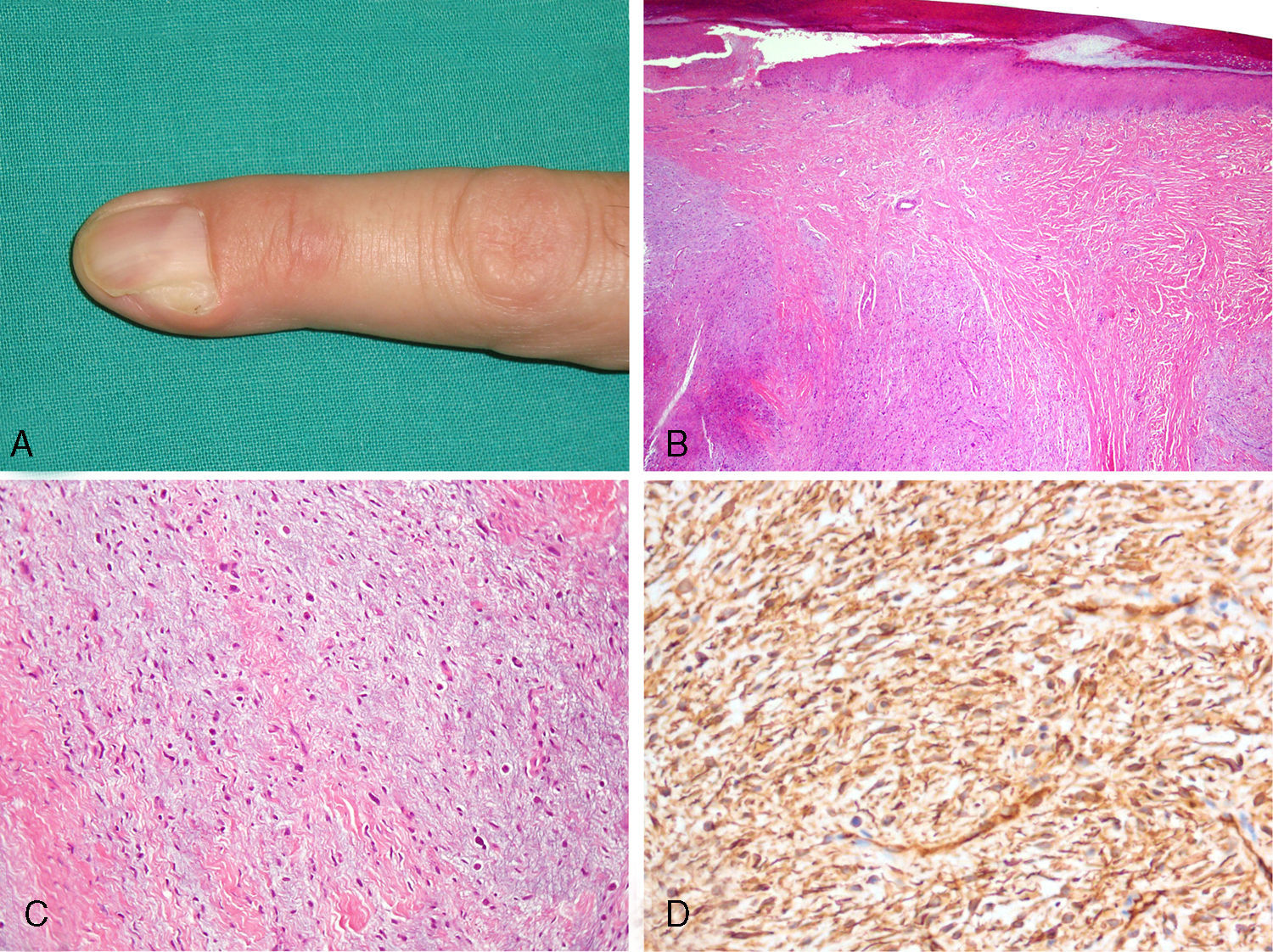
A) Nódulo subcutáneo en el segundo dedo de la mano izquierda, que deforma parcialmente la matriz ungueal. B) Tumoración dérmica constituida por áreas fibromixoides y mixoides dispuestas de forma alternante. C) Células fusiformes incluidas en un estroma mixoide. También se observan numerosos mastocitos de núcleo redondo y citoplasma más amplio (hematoxilina-eosina X100). D) Positividad citoplasmática difusa para CD34 en las células neoplásicas (X200).
El estudio histológico de la lesión, después de realizar su exéresis simple con curetaje del fondo quirúrgico, mostraba una epidermis con hiperqueratosis ortoqueratósica leve y una neoplasia dérmica mal delimitada (fig. 1B), formada por una población de células fusiformes inmersas en un estroma laxo constituido por áreas mixoides y fibromixoides, de igual densidad celular, dispuestas de forma alternante sin un patrón arquitectural definido (fig. 1C). También existían numerosos mastocitos (positivos para CD117). La tinción con azul alcián puso de manifiesto la presencia de mucopolisacáridos ácidos que predominaban en las áreas menos fibrosas. El componente celular no mostraba atipia nuclear ni mitosis. Las células neoplásicas presentaban, de forma difusa en todo el espesor de la tumoración, positividad intensa para CD34 (fig. 1D) y CD99, con un índice Ki-67 bajo (< 5%). Las tinciones para antígeno de membrana epitelial (EMA) y proteína S100 fueron negativas. Con estos hallazgos se estableció el diagnóstico de fibromixoma acral superficial (FAS). Los márgenes laterales y profundos de la pieza resecada estaban afectados por lo que se consideró inicialmente la ampliación quirúrgica. Sin embargo, el paciente rechazó la reintervención. Ha recibido revisiones periódicas y permanece asintomático después de 6 meses. El estudio mediante resonancia magnética (RM) realizado a los 4 meses de seguimiento no mostró signos de persistencia tumoral.
El FAS es una neoplasia descrita en 2001 por Fetsch et al., quienes presentaron una serie de 37 casos después de realizar una revisión sistemática de 280 tumores acrales fibrohistiocitarios diagnosticados durante tres décadas como fibroma, mixoma, fibromixoma, mixolipoma, dermatofibroma, histiocitomas fibroso o angiofibroma, entre otros1. Posteriormente han sido publicadas varias series de FAS y algunos casos aislados2–6. Se trata de una lesión benigna, sin casos conocidos de malignización y/o metástasis1,2. Hay pocos casos descritos de recidiva y siempre asociados a una extirpación incompleta3,4. Existen alrededor de 100 casos en la literatura y algunos autores consideran que es un tumor infradiagnosticado.
Clínicamente muestra preferencia por el sexo masculino (2:1) y se ha observado en pacientes con edades comprendidas entre los 14 y los 75 años, con una mediana de 46 años1. Suele presentarse como un nódulo de consistencia sólida, con un crecimiento lento e indoloro, lo que motiva un retraso en la consulta. Por orden de frecuencia sus principales localizaciones son los dedos de los pies, principalmente el primer dedo, los dedos de las manos y, más raramente, las palmas y las plantas1,3,4. La uña está afectada en un 50% de casos con hiperqueratosis u onicolisis que se acompaña a veces de dolor a la compresión. Es rara la erosión del hueso subyacente aunque sí está descrita7.
Histológicamente, a diferencia de lo observado en nuestro caso, se trata de una tumoración dérmica o subcutánea bien delimitada, no encapsulada, que presenta un aumento en su vascularización1–5. También ha sido descrito en algunos casos de FAS la presencia de células multinucleadas estromales, áreas de necrosis, áreas lipomatosas y signos epidérmicos de infección viral1–5,8. Este último hallazgo ha llevado a sugerir la implicación del virus del papiloma humano en la etiopatogenia del FAS4. En el estudio inmunohistoquímico es característica la expresión de CD34, aunque existen casos negativos para este marcador9. También es habitual la expresión de CD10, CD99, EMA y nestina. Dada la positividad para estos marcadores, algunos autores postulan que este tumor tiene su origen en células madre mesenquimales residentes en zonas acras1,8,10. Son negativas, de forma casi constante, las inmunotinciones para S100, actina, desmina, citoqueratina, apolipoproteína D y HMB451,2,4.
La inmunorreactividad para CD34 obliga descartar otras entidades CD34 positivas (tabla 1), sobre todo el dermatofibrosarcoma protuberans mixoide (DFSPM). Clásicamente se consideraba que el DFSPM era más frecuente en zonas acras que otras formas de dermatofibrosarcoma protuberans. Sin embargo, esta diferencia en el patrón de distribución podría deberse a que muchos de los casos descritos de DFSPM correspondían en realidad a FAS. El DFSPM expresa apolipoproteína D y muestra áreas con patrón estoriforme e infiltración subcutánea característica. La presencia de la traslocación (17;22) permite confirmar el diagnóstico. También deberán considerarse en el diagnóstico diferencial lesiones mixoides de células fusiformes, tanto benignas (neurofibroma mixoide, angiomixoma superficial, quiste mucoide) como malignas (DFSPM, sarcoma fibroblástico mixoinflamatorio acral, sarcoma fibromixoide de bajo grado, mixofibrosarcoma) y otras neoplasias acrales como el perineuroma esclerosante, el fibroma periungueal, el fibroqueratoma digital y el fibroma digital celular. Esta última entidad está constituida por células fusiformes CD34 positivas y se diferencia del FAS por ser menos mixoide y más celular, aunque algunos autores postulan que se trata de la misma entidad11.
Tumores fusocelulares con expresión de CD34
| Entidad (frecuencia CD34+) | Hallazgos característicos | Inmunohistoquímica |
| Muy frecuente | ||
| Dermatofibrosarcoma protuberans | Traslocación (17;22)Patrón estoriforme e infiltración subcutánea digitiforme | Apo D, actina y vimentina positivasS-100, HMB-45, citoqueratinas y factor XIIIa negativos |
| Tumor fibroso solitario | Tumor benigno muy infrecuente en la piel, donde suele localizarse en cabeza y cuello. Gran variabilidad histológica | CD 99 y vimentina positivosS-100 y marcadores musculares negativos. |
| Fibroma esclerótico | Similar al tumor fibroso solitario | |
| Tumores de la vaina nerviosa (neurofibroma, neuroma) | Núcleos ondulados | S-100 positiva |
| Lipoma de células fusiformes | S-100 positiva | |
| Fibromixoma acral superficial (FAS) | Áreas fibromixoides, aumento en la vascularización y presencia de mastocitos. | EMA, CD99, CD10 y nestina positivos.S-100, Apo D, citoqueratina, vimentina, y desmina negativas. |
| Fibroma digital celular | Similar al FASMenos mixoide y más celular | Similar al FAS |
| Variable/ocasional | ||
| Sarcoma epitelioide | Nódulos o grupos de células epitelioides rodeando una zona necrótica central | Citoqueratinas, EMA y vimentina positivas |
| Sarcoma fibroblástico mixoinflamatorio acral | Patrón de crecimiento infiltrativo, importante componente inflamatorio, estroma mixoide o hialino con tres poblaciones de células tumorales (multinucleadas a tipo Reed-Sternberg, epitelioideas y fusiformes) | Vimentina positiva, positividad ocasional para CD34 y CD68Negatividad para EMA y S-100 |
| Mixofibrosarcoma de bajo grado | Gran pleomorfismo celular, numerosas mitosis atípicas, vasos capilares curvilíneos prominentes | Vimentina y actina positivas,CD34 ocasionalmente positivoDesmina y EMA negativas |
| Angiosarcoma | Presencia de luces vasculares con un patrón infiltrativo, extravasación hemática y depósitos de hemosiderina | Positividad para marcadores vasculares sanguíneos y linfáticos (F-VIII-RA, D2-40, CD31) |
| Dermatofibroma | CD34 excepcionalmente positivoFactor XIIIa positivo | |
El tratamiento del FAS consiste en la resección quirúrgica con márgenes libres de tumor1,3. Dada la capacidad de recidiva, se recomienda realizar un seguimiento estrecho a estos pacientes tras la extirpación, sobre todo en casos como el nuestro con márgenes de resección afectos1,3.
En conclusión, dermatólogos y dermatopatólogos deben considerar esta entidad en el diagnóstico diferencial de los tumores fibrohistiocitarios acrales. Un diagnóstico precoz y una resección completa son fundamentales para evitar posteriores recidivas.


