


INTRODUCCION
La micosis fungoide, junto con su variante leucémica y eritrodérmica, el síndrome de Sézary es el más frecuente de los linfomas cutáneos de células T (LCCT). Tiende a permanecer visible en la piel durante años y sigue una evolución en estadios (mancha, placa y tumor), pudiendo en algunos pacientes dejar la piel y diseminarse a ganglios linfáticos y vísceras y acabar con su vida1-3.
Hay varios trabajos de seguimiento a grandes grupos con esta enfermedad, la mayoría de ellos provenientes de Estados Unidos4-13, aunque también los hay europeos14-16. En estos trabajos se apuntan algunos factores que influyen en la supervivencia: extensión y tipo de lesión cutánea, estadio T, afectación linfática o visceral, células de Sézary circulantes, edad, lacticodeshidrogenasa (LDH), receptor soluble de interleucina-2 (IL-2), presencia de un clon específico en sangre periférica, grosor del infiltrado tumoral, etc. Son menos los artículos que estudian los factores de progresión y la frecuencia de esta progresión10-14. A pesar de todo, estas variables pronósticas permanecen menos definidas que en otros tipos de linfomas. Por otra parte, la comparación entre estas series es a menudo difícil por los diferentes criterios de inclusión empleados y en ocasiones por el uso como sinónimos de micosis fungoide y LCCT.
En este trabajo se pretenden describir las características clinicoepidemiológicas, de una serie mixta, histórica y concurrente, de pacientes con micosis fungoide/síndrome de Sézary, determinar la existencia de posibles factores de riesgo (clínicos y analíticos) de progresión neoplásica en estos pacientes hacia una fase más agresiva de la enfermedad y determinar la mortalidad y supervivencia y los factores predictivos asociados en los sujetos de estudio.
MATERIAL Y MÉTODOS
Hemos estudiado 112 pacientes diagnosticados de micosis fungoide/síndrome de Sézary en un área sanitaria por criterios clínicos y patológicos generalmente aceptados17. Se han recogido los datos de pacientes históricos, desde 1976 a 1998, y de los nuevos diagnósticos desde 1998 hasta el año 1999. De ellos, se han excluido posteriormente 12 casos dudosos, quedando un total de 100 pacientes, que han sido seguidos un mínimo de 1 año (1-23 años). Se ha considerado el momento del diagnóstico el momento en que se ha realizado la primera biopsia, aunque también se ha tenido en cuenta el momento de inicio de la enfermedad en función de los datos clínicos.
Se define la remisión completa (RC) como la desaparición total de todas las lesiones y completa mantenida cuando persiste al menos 2 años sin recaída. No se hace distinción entre respuesta parcial y no respuesta. El estadio de la enfermedad se ha determinado de acuerdo con el sistema TNM18. Se define progresión cuando la enfermedad evoluciona a un estadio TNM superior. Se estudia tanto la supervivencia y mortalidad específica (analizando aquí sólo los pacientes cuyo fallecimiento es directamente imputable a la micosis fungoide) como la global (fallecimiento por cualquier causa).
Se han considerado pacientes perdidos en el seguimiento aquellos que no sabemos su estado vital en el momento de cerrarse el estudio.
Se han analizado las siguientes variables: tipo de lesión cutánea (T1, T2, T3, T4), adenopatías (N0, N1, N2, N3), afectación de sangre periférica (células de Sézary circulantes: punto de corte > 10 %), estadificación TNM, edad (punto de corte 60 años), sexo, respuesta completa al tratamiento inicial, β2-microglobulina (punto de corte 2,4 mg/l), factor de necrosis tumoral α (TNF-α) (punto de corte 28 pg/ml), LDH (punto de corte 300 U/dl), CD25 o receptor soluble de la IL-2 (770 U/ml), eosinofilia en sangre periférica (punto de corte 600 eosinófilos/ml), CD7 en sangre periférica (punto corte 20 %), presencia de reordenamiento genético positivo o negativo en piel y sangre periférica, año de diagnóstico y retraso del comienzo-diagnóstico.
Análisis estadístico
El análisis se desglosa en cinco apartados: descriptivos generales y análisis de respuesta al tratamiento, progresión de la enfermedad, general de mortalidad por linfoma y análisis de supervivencia para la mortalidad por linfoma.
Estadística
En general, la comparación entre dos variables cualitativas se ha realizado mediante test de chi cuadrado (χ2) de Pearson. En algunos casos de variables dicotómicas y en tablas con efectivos esperados menores de 5 se ha empleado la prueba exacta de Fisher. En variables ordinales se ha estimado la asociación lineal mediante test de Mantel-Haentzel. La comparación entre dos medias se ha realizado mediante test de la t de Student, con cálculo de intervalos de confianza (IC) de la diferencia (asumiendo un error alfa del 5 %). El análisis ajustado de la asociación de una variable dependiente dicotómica con otras independientes se ha efectuado por regresión logística, siguiendo el método por pasos y estimación de las medias de asociación de razón de ocurrencia odds ratio (OR). Para el análisis de supervivencia se ha empleado el método actuarial y de Kaplan-Meyer. Se ha realizado un análisis de supervivencia global y otro estratificado según varias variables. La diferencia de supervivencia entre los estratos se ha comparado mediante los tests estadísticos de rango logarítmico o log rank, Breslow y Tarone-Ware. Finalmente, se ha realizado un análisis de supervivencia ajustado por posibles variables predictoras mediante riesgos proporcionales o regresión de Cox.
RESULTADOS
Características clínicas generales
La muestra de 100 pacientes está compuesta por el 59 % de hombres y un 41 % de mujeres. La edad media de comienzo de la enfermedad fue de 48,2 años (IC de 44,3 a 52,1 años), con un rango de 14 a 91 años. La edad media de diagnóstico de la enfermedad fue de 54,4 años (IC de 50,9 a 57,9 años) variando entre los 15 y los 91 años. Hubo 8 pacientes con un inicio del proceso antes de los 20 años, todos ellos en estadios IA-IIA.
La casuística se distribuye entre los años 1976 y 1999. El tiempo medio transcurrido entre el comienzo de la enfermedad y el diagnóstico osciló entre un mes y 35 años con una media de 6,2 años (IC de 4,6 a 7,7 años). Esta demora fue mayor en los estadios iniciales IA-IIA. El tiempo medio transcurrido entre comienzo de la enfermedad y diagnóstico se ha reducido sensiblemente en la última década. Hasta 1990, este tiempo de retraso tuvo una media de 8 años, mientras que a partir de 1990 este tiempo ha sido de 1, 2 años (IC de 8,8 a 4,8). Estas diferencias resultaron estadísticamente significativas (p < 0,01). El IC indica que en la actualidad el diagnóstico se produce entre 4,8 y 8,8 años antes que en la década anterior.
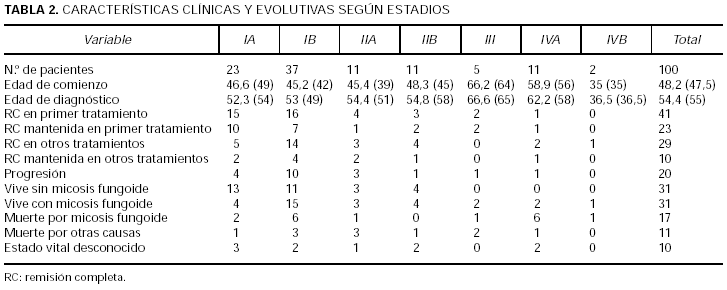
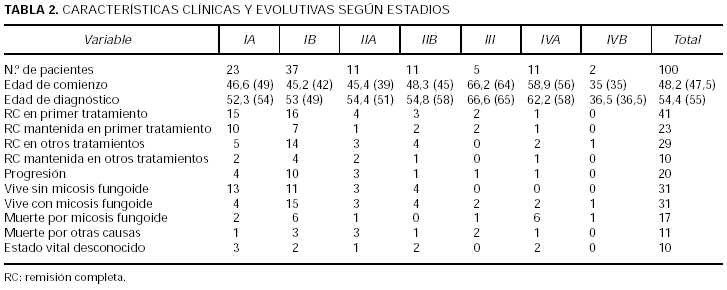
En la tabla 1 se presentan los estadios al diagnóstico y en la tabla 2 las características clínicas y evolutivas según los distintos estadios. La mayor parte de los pacientes incluidos en este estudio corresponden a estadios iniciales IA o IB (60 %), mientras que sólo el 7 % se presentaron con tumores o eritrodermia sin afectación extracutánea. Globalmente, el 67 % de los pacientes tenían únicamente afectación cutánea. En total, 13 pacientes se presentaron con tumores (cinco con afectación sólo cutánea) y un 11 % con eritrodermia (dos afectación exclusivamente cutánea). El 21 % presentó adenopatías palpables no confirmadas histológicamente y el 13 % afectación ganglionar o visceral demostrable (los 2 casos con afectación visceral correspondieron a médula ósea) (tabla 1).

Análisis de respuesta al tratamiento
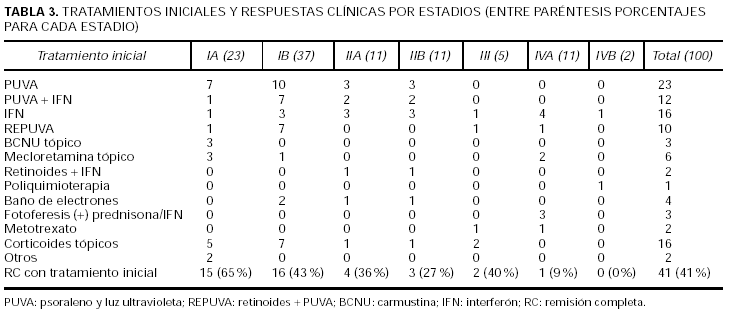
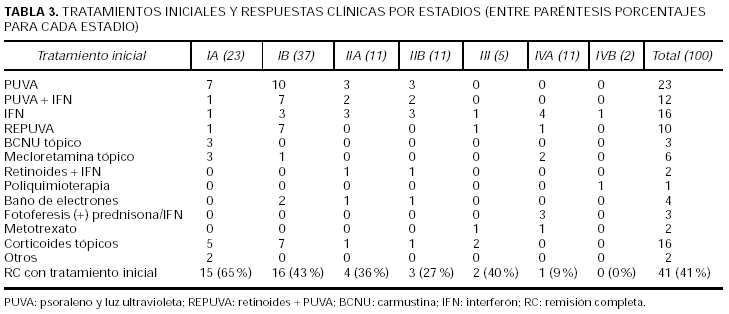
La tabla 3 presenta las terapéuticas iniciales por estadios y el índice de RC con dicho tratamiento. Los tratamientos iniciales son similares a otras series europeas14, si bien difieren de las americanas por el menor uso de tratamientos agresivos como radioterapia con baño de electrones o poliquimioterapia12. El tratamiento más utilizado en estadios iniciales es el psoraleno y luz ultravioleta (PUVA) seguida de corticoides tópicos, interferón(IFN), PUVA + IFN y carmustina (BCNU) o mecloretamina tópicos. El tratamiento inicial consiguió la RC en 41 casos. De ellos, 23 pacientes han conseguido RC duraderas. El resto de los pacientes sólo 10 consiguieron remisiones duraderas con los tratamientos sucesivos.
El estudio estadístico mostró que el 65 % de los estadios T1 remitieron con el primer tratamiento, mientras que este porcentaje fue sólo del 18,8 % entre los que presentaron estadio T4. Estas diferencias fueron estadísticamente significativas (p = 0,02) (χ2 de Pearson) y además se describe una tendencia lineal (a mayor estadio T menor porcentaje de respuesta) también significativa (p = 0,003). Lo mismo sucedió con el estadio N: remitieron más los pacientes N0 que N3 (p = 0,018), con una tendencia lineal significativa (p = 0,005).
El análisis según estadios indica que la prevalencia de remisión al primer tratamiento no fue homogénea por grupos de estadio (p = 0,03). El porcentaje de remisiones fue superior en los grupos IA al diagnóstico (68,18 %). No se mostró asociación lineal, probablemente debido al alto índice de remisión completa de los pacientes en estadio III (2/5), posiblemente dado lo reducido del grupo. Se ha realizado un análisis ajustado de respuesta al tratamiento, utilizándose 4 modelos distintos de regresión logística, para así poder comparar un mayor número de variables. La remisión se asoció al estadio N al diagnóstico comparando N3 con N1 y N0 (p = 0,03; OR = 11); no existieron diferencias entre N1 y N0. En cuanto al estadio T sólo hubo diferencias comparando T1 frente a T2-4. Además, la remisión se asoció al estadio, comparando todos con IA (excepto III frente a IA p = 0,25). En ningún modelo se encontró asociación estadística con edad, sexo y retraso en el diagnóstico.
Análisis de la progresión
Globalmente, un 20 % de los pacientes progresaron a un estadio mayor mientras que un 35 % prosiguió un curso fluctuante pero sin progresar. En la tabla 4 se desglosa la progresión por estadios. Se ha analizado la variable dependiente progresión con otras posibles variables predictoras: estadio T al diagnóstico, estadio N al diagnóstico, sexo, remisión con el primer tratamiento, tiempo de retraso del diagnóstico, LDH y eosinofilia. En los primeros análisis univariantes crudos la progresión sólo se asoció a la ausencia de remisión al primer tratamiento.

Se realizó un análisis ajustado por regresión logística utilizando, como en el apartado anterior, 4 modelos. No se ha incorporado el resto de los parámetros analíticos (excepto LDH y eosinofilia tomados al inicio del cuadro) por dos motivos: no existe una correlación temporal clara entre la determinación y la ocurrencia de progresión y no se puede saturar de variables una regresión logística.
En los 4 modelos la progresión se asoció a la falta de remisión con el primer tratamiento. Además, también se asoció al estadio T1-T2 frente a T2-T4 (p = 0,005), es decir, progresaron más los que menor estadio presentaron. Sin embargo, no hubo diferencia al comparar T1 frente al resto de los estadios T por separado. Tampoco hubo asociación en las diversas combinaciones del estadio N ni de los estadios TNM, como tampoco la hubo con edad, sexo o retraso al diagnóstico.
Análisis de la mortalidad por linfoma
En la tabla 2 se observa la distribución de los pacientes según su estado vital. Globalmente ha fallecido el 28 % de los pacientes, 17 pacientes por linfoma y 11 por otras causas. Cinco se encuentran vivos, 62 pacientes libres de enfermedad y otros 31 con micosis fungoide. En un 10 % de los pacientes se desconoce su estado actual. Para saber si la pérdida de esos pacientes puede haberse debido a algunos factores intrínsecos de la enfermedad como una mayor gravedad o progresión que hayan condicionado su supervivencia o mortalidad se ha realizado un estudio estadístico comparando el grupo con estado vital desconocido y conocido respecto a variables de interés. Se han comparado con respecto a estadio T al diagnóstico, estadio N, progresión, respuesta al primer tratamiento y edad, y no se encontraron diferencias estadísticamente significativas entre ambos grupos.
Se ha comparado la variable "mortalidad por linfoma" con otras posibles variables predictoras. En todos los grupos se ha comparado mortalidad por linfoma con respecto a pacientes vivos o muertos por otras causas.
Realizando un análisis univariante crudo no se observó asociación con el estadio T al diagnóstico (p = 0,317). La mortalidad por linfoma en el grupo T1 al diagnóstico fue del 5 %, mientras que para T2 fue del 20,8 %. Sin embargo, estas diferencias no fueron significativas (prueba de la χ2 y estadístico exacto de Fisher). Cuando comparamos mortalidad por linfoma para el grupo T1 frente a T2-4 la mortalidad por linfoma en el grupo T1 al diagnóstico fue de 5 %, mientras que para T2-T4 fue de 21,7 %. Sin embargo, estas diferencias tampoco fueron significativas (p = 0,09).
El análisis univariante crudo según el estadio N al diagnóstico mostró una p < 0,05 (p = 0,001), con lo cual esta variable influía en la mortalidad. Como en el apartado anterior, el análisis se ha estratificado en subgrupos. Comparando N0 frente a N1 al diagnóstico se observa una mortalidad por linfoma en el grupo N0 del 13,1 %, mientras que para N1 fue de 11,1 %. Sin embargo, estas diferencias no fueron significativas (p = 1,00). Comparando N0-1 frente a N3: la mortalidad por linfoma en el grupo N0-1 al diagnóstico fue de 12,6 %, mientras que para N3 fue de 60 %. Estas diferencias fueron estadísticamente significativas (p = 0,002). Analizando el estadio TN al diagnóstico se observa que el estadio con mayor mortalidad por linfoma fue el IVA (66,6 %). Estas diferencias son estadísticamente significativas (p = 0,001). El análisis univariante también mostró asociación a la mortalidad con la progresión de la micosis fungoide (p < 0,001), falta de remisión al primer tratamiento (p = 0,006), así como con datos de laboratorio tomados en diferentes momentos de la evolución: eosinofilia > 600 (p = 0,009), células de Sézary circulantes > 10 % (p = 0,0007), b2-microglobulina > 2,4 (p = 0,011) y LDH > 300 (p = 0,004). Además, también mostró asociación la cifra de CD7 negativo en sangre periférica superior al 20 % de linfocitos circulantes (p = 0,0011; OR = 30); esta variable no se incluyó en el estudio ajustado por regresión logística. La mortalidad por micosis fungoide no se asoció a las cifras de receptor soluble de IL-2 y TNF-α.
En el análisis ajustado por regresión logística la mortalidad por linfoma se ha asociado a la progresión (p = 0,001; OR = 65,2; comparando los que progresaron, frente a los que no progresaron); a la edad (p = 0,02; OR = 10,7; comparando los mayores de 60 frente al resto), al estadio N al diagnóstico (p = 0,005; OR = 41,4; comparando estadio 3 con estadio 0) a y la eosinofilia (p = 0,04; OR = 7,9; comparando los que tenían valores de más de 600 frente al resto). No hubo asociación al retraso comienzo-diagnóstico y las demás variables perdieron su asociación. Finalmente, se ha calculado la densidad de incidencia para la mortalidad por linfoma que es 0,023 casos por años-1 y para la mortalidad total (0,038 casos por año-1). Esto quiere decir que de 100 pacientes seguidos 10 años, 23 fallecen por linfoma y 38 teniendo en cuenta la mortalidad por cualquier causa.
Análisis de supervivencia para la mortalidad por linfoma
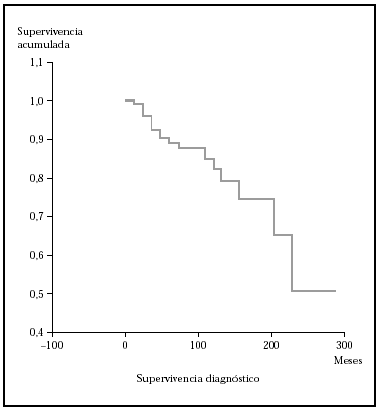
En este apartado se analiza la probabilidad de supervivencia si sólo se tiene en cuenta la mortalidad por linfoma. En nuestra serie, la probabilidad acumulada de supervivencia en el tiempo (de no morir por linfoma) es del 87,5 a los 5 años, de1 79,12 % a los 10 años y del 74,17 a los 15 años. La mediana para nuestros pacientes fue mayor de 276 meses (el 50 % de los sujetos no han muerto por linfoma pasado este tiempo). En la figura 1 se muestra el gráfico de supervivencia para la mortalidad por linfoma.
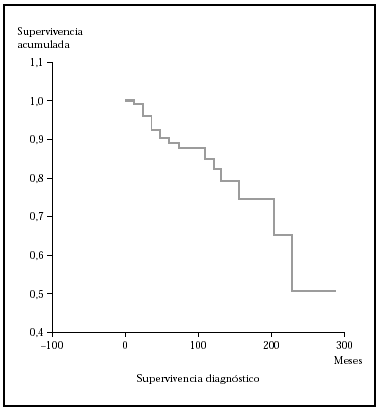
Fig. 1.--Curva de Kaplan-Meier indicando la supervivencia específica para la mortalidad por linfoma desde el diagnóstico. A los 276 meses todavía no han fallecido el 50 % de los pacientes.
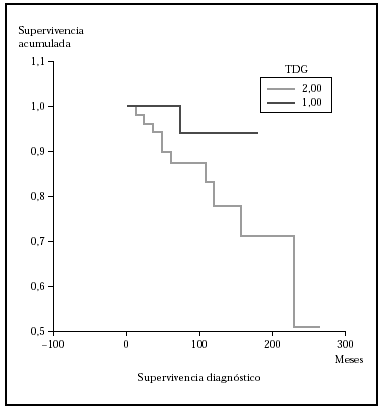
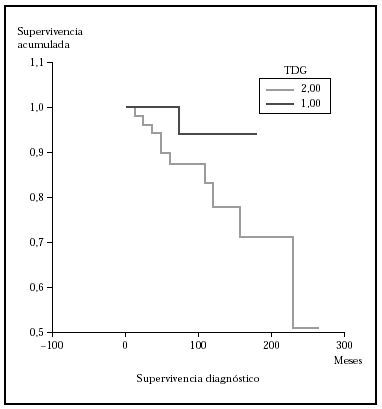
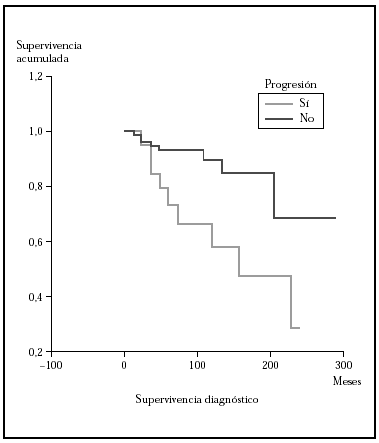
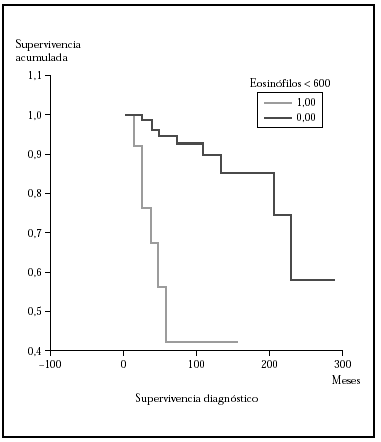
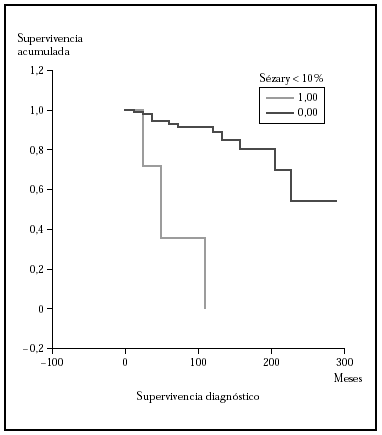
Posteriormente se ha repetido el análisis de supervivencia, pero estratificado por algunas variables independientes o pronósticas. Se han comparado el grupo T1 frente a T2 (mediana > 168 frente > 252), y T1 frente a T2-4 (mediana > 168 frente a 27) y aunque hay una mayor supervivencia en T1 las diferencias no fueron estadísticamente significativas (fig. 2). El análisis estratificado por estadio N al diagnóstico no mostró diferencias significativas comparando N0 frente a N1-3 (mediana > 252 meses frente 203), pero sí al comparar N0-1 frente a N3 (mediana > de 276 meses frente a 102) (fig. 3). Se han encontrado diferencias significativas en la progresión (mediana de supervivencia entre los que progresaron de 153 meses frente a una mediana superior a 275 meses entre los que no lo hicieron) (fig. 4) y en la respuesta al primer tratamiento (entre los que remitieron la mediana fue superior a los 252 meses y entre los que no de 201,9 meses). Además, la supervivencia fue mayor en los pacientes con eosinofilia < 600 (mediana > 276 frente a 53) y células de Sézary circulantes < 10 % (mediana > 276 frente a 43 meses) (figs. 5 y 6). En todos los análisis donde se han encontrado diferencias significativas, éstas lo han sido según los tres tests empleados (log rank, Breslow y Tarone-Ware).
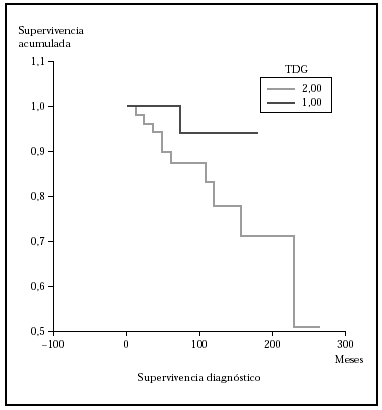
Fig. 2.--Supervivencia para la mortalidad por linfoma en pacientes con estadio T al diagnóstico (TDG) T1 frente a T2 (método Kaplan-Meier). Aunque se observa diferencia en la supervivencia, ésta no fue estadísticamente significativa.
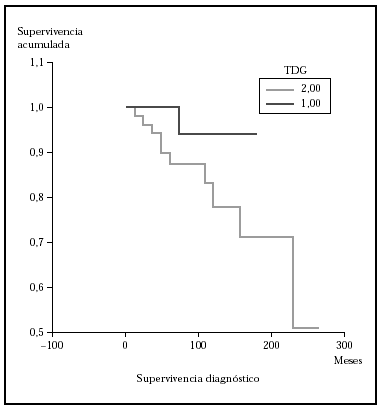
Fig. 3.--Supervivencia para la mortalidad por linfoma en pacientes con estadio N0-1 al diagnóstico frente a N3 (ND013) (método Kaplan-Meier). La supervivencia fue mayor en el grupo N0-1 y fueron estadísticamente significativas, p = 0,0003 (test log rank); p = 0,001 (Breslow); p = 0,0005 (Tarone-Ware).
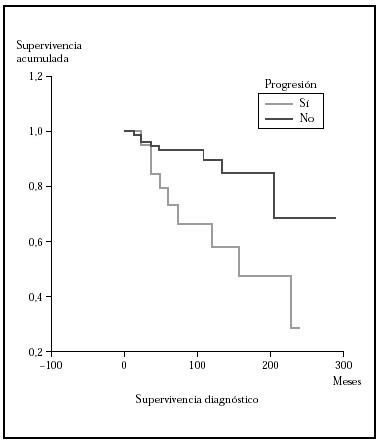
Fig. 4.--Comparación por el método Kaplan-Meier de la supervivencia para la mortalidad por linfoma entre los pacientes que progresan a un estadio mayor frente a los que no lo hacen. La mediana de supervivencia para los pacientes que progresaron fue de 153 meses mientras que los que no lo hicieron no la habían alcanzado a los 275. Estas diferencias fueron estadísticamente significativas.
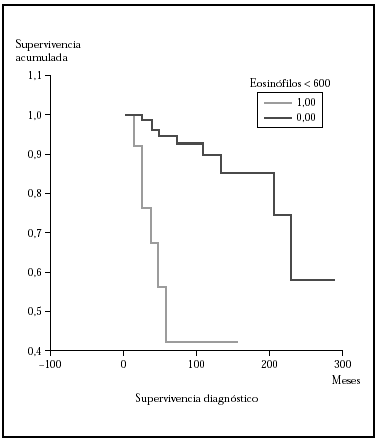
Fig. 5.--Curva de supervivencia para la mortalidad por linfoma donde se muestra que la supervivencia fue mayor en el grupo con < 600 eosinófilos/ml. Mediana mayor de 276 meses frente a 53 meses. Estas diferencias fueron estadísticamente significativas (p > 0,00001) con los tres test (log rank, Breslow, Tarone-Ware).
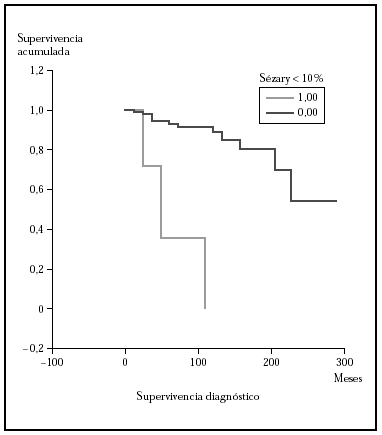
Fig. 6.--Análisis estratificado de supervivencia para la mortalidad por linfoma según células de Sézary circulantes. Las curvas de Kaplan-Meier muestran una diferencia significativa (p > 0,00001) a favor del grupo con una sezaremia < 10 %.
Finalmente se ha realizado un análisis ajustado de supervivencia en el cual se han considerado simultáneamente todas las variables, introduciendo además la edad y el retraso entre comienzo de la enfermedad y diagnóstico. Las variables asociadas a una menor supervivencia (considerando el linfoma como suceso mortal final) fueron la progresión de la enfermedad (p = 0,007; OR = 5,06), la edad mayor de 60 años (p = 0,04; OR = 3,9), las células de Sézary circulantes mayor del 10 % (p = 0,002; OR = 7,5), y la eosinofilia mayor de 600 (p = 0,006; OR = 6,03). El estadio N (0-1 frente 3) fue casi significativo (p = 0,055). El retraso en el diagnóstico y la remisión al primer tratamiento perdieron relevancia en este análisis ajustado.
DISCUSION
En este trabajo se han analizado las principales características clínicas, analíticas, evolutivas, supervivencia y mortalidad de 100 pacientes con micosis fungoide/síndrome de Sézary diagnosticados entre 1976 y 1999. Además, se han analizado numerosas variables implicadas en la progresión de la enfermedad, supervivencia, mortalidad y respuesta al tratamiento.
En todos los estudios de pacientes con micosis fungoide se encuentra presente el sesgo de inclusión de la denominada parapsoriasis en placas y su diferenciación de micosis fungoide en estadios precoces. El término hace referencia a pacientes con manchas o placas apenas infiltradas, pero sin hallazgos histológicos de micosis fungoide. La controversia más reciente la protagonizan Ackerman y Burg19-21 ya que el primero considera las parapsoriasis como presentaciones clínicas de micosis fungoide, y el segundo como un linfoma cutáneo abortivo dentro del proceso multifactorial y en distintos pasos de la carcinogénesis. Los estudios de clonalidad no permiten una distinción clara entre micosis fungoide precoz y parapsoriasis ya que ambas pueden reordenar en piel y sangre periférica22-24. La histología y el aspecto clínico en esta fase, en numerosas ocasiones tampoco son delimitorias. Además, las micoides fungoide precoces tienen un riesgo bajo de desarrollar formas agresivas de la enfermedad12-14 por lo que la evolución a largo plazo en algunos pacientes será la única manera de saber a posteriori qué pacientes progresan, pero esta técnica es poco aceptable. Así pues, algunos autores ponen en duda la utilidad del término parapsoriasis en placas y la cuestión realmente importante sería si es verdaderamente o no una micosis fungoide14. Será necesario buscar nuevos modos de definir a priori qué pacientes tienen riesgo de progresión. Nosotros hemos incluido pacientes con síntomas sugerentes de micosis fungoide en los que hay correlación clinicopatológica, y excluimos 12 casos dudosos de los 112 pacientes diagnosticados inicialmente.
En nuestra serie la relación varón/mujer fue 1,44. Esto supone un ligero predominio de los hombres, al igual que en otros trabajos, que oscila entre 1,3-2,2/19,13,25,26. La edad media de comienzo (48 años) y de diagnóstico (54) es congruente con otros estudios, aunque se obtuvo un mayor porcentaje (8 %) de paciente jóvenes (< 20 años). De ellos, cinco están en el momento actual en remisión completa y tres viven con enfermedad limitada (IA) después de un tiempo de evolución entre 5 y 49 años (media, 18,65). Algunos trabajos han apuntado que en las micosis fungoide de comienzo en una edad temprana no existen diferencias estadísticamente significativas respecto al comienzo en edad adulta27.
Llama la atención la reducción del tiempo comienzo-diagnóstico. Se ha pasado de una media de 8 años antes de 1990 a 1,2 en la actualidad, diagnosticándose entre 4,8 y 8,8 años antes. A pesar de realizarse un diagnóstico más temprano no ha disminuido la edad media al diagnóstico, 53,3 años antes de 1990 y 55 después de 1990. Sí que se ha producido un mayor diagnóstico de pacientes en estadio IA (25 % antes de 1990 frente a 15 % posterior a esa fecha) y un menor diagnóstico de estadios IVA (10 % frente a 15 %), pero esas cifras no son estadísticamente significativas (p = 0,157). El número de pacientes diagnosticados es considerablemente mayor en la última década que en años anteriores (76/100 pacientes han sido diagnosticados a partir de 1989 pero, posiblemente hayan influido errores en el registro de algunos pacientes a finales de 1970). No obstante, no existen diferencias estadísticamente significativas en cuanto a mortalidad según la fecha de diagnóstico. Esto es lógico si se considera que, a pesar de la disminución del tiempo comienzo-diagnóstico, no ha disminuido la edad media al diagnóstico, ni se diagnostica de forma estadísticamente significativa en estadios más precoces; estos hechos también han sido apuntados por otros autores. Además, hay que tener en cuenta que, a pesar de los avances terapéuticos todavía no se dispone de un tratamiento curativo para esta enfermedad.
En algunos trabajos se ha reflejado un aumento de la incidencia de esta enfermedad25,28. En nuestros casos, se ha producido un aumento del número de pacientes hasta 1989, manteniéndose posteriormente más o menos constante; esta tendencia es la misma que la mostrada en dos recientes trabajos26,29. Esto, y el hecho apuntado anteriormente de una menor demora diagnóstica, puede deberse a un mayor conocimiento de la enfermedad, una mejor definición de los criterios diagnósticos, un mejor acceso a los sistemas de salud y un registro y seguimiento de estos pacientes más adecuados. También ha influido notablemente, sobre todo en las series americanas, la aparición a finales de 1970 de artículos que delineaban los criterios patológicos de la micosis fungoide en estadios precoces, incluyendo muchas de las anteriormente denominadas parapsoriasis en placas. Como en otras series9,13,14, la mayoría de los pacientes en el momento del diagnóstico tenían únicamente afectación cutánea (67 %). A este porcentaje podría sumársele un 20 % con adenopatías palpables, lo que nos dejaría que sólo el 13 % tenía afectación ganglionar o visceral en el momento del diagnóstico.
Los tratamientos iniciales más utilizados han sido aquellos que van dirigidos directamente contra el lugar en el que asienta la enfermedad, es decir, la piel como corticoides tópicos, PUVA, BCNU/mecloretamina tópica u otros. Además, en cierto número de pacientes se han utilizado modificadores de la respuesta inmunitaria como el IFN en monoterapia o en combinación. Se han reservado tratamientos más agresivos como radioterapia y mono o poliquimioterapia para no respuestas a los tratamientos anteriores o estadios avanzados. Con estos tratamientos se ha conseguido una RC del 41 %. De ellos, más de la mitad (23 pacientes) han conseguido RC duraderas (más de 2 años), lo cual es superior a otras series europeas14. Además si se añaden las RC duraderas con otros tratamientos, éstas se elevan a la tercera parte del total de los pacientes. De dichas RC duraderas casi la mitad (15 pacientes) han sido superiores a 5 años con el primer tratamiento; de ellos sólo dos han recidivado posteriormente y se encuentran en el momento actual vivos pero con enfermedad, sin haber fallecido ninguno de ellos. Además, hay 4 pacientes con RC tras el primer tratamiento superiores a los 10 años, lo que en la práctica puede significar "curaciones potenciales". En nuestro estudio han respondido de forma significativamente superior los pacientes con menor estadio T al diagnóstico y con menor estadio N y los pacientes diagnosticados en estadio IA. No influyen en una mejor respuesta la edad, sexo y retraso en el diagnóstico. Además, la no RC al primer tratamiento ha demostrado ser una variable independiente predictora de progresión. La no RC al primer tratamiento fue una variable predictora de mortalidad por linfoma; en el análisis univariante, fallecieron el 28 % de los pacientes que no respondieron al primer tratamiento frente al 5,4 % de los que sí respondieron (p = 0,006). Sin embargo, y a pesar de perderse la significación estadística en el estudio ajustado (multivariante), esas cifras tienen significación biológica. Esto se comprueba en que tanto en el análisis de supervivencia para mortalidad global como por linfoma volvieron a ser variables significativas en el análisis univariante. Además, el hecho de que respondan mejor los pacientes en estadios precoces y de que progresen más los que no responden nos hace pensar que, con un mayor número de pacientes y años de seguimiento, podría comportarse como una variable pronóstica independiente.
No se han comparado la eficacia e influencia de los distintos tipos de tratamiento en la micosis fungoide debido al número elevado de tratamientos empleados, a que no haya unos criterios tipificados para aplicar un tratamiento determinado en una situación determinada y a que es posible que el grado de afectación de la enfermedad haya influido en el tratamiento elegido. No obstante son varios los artículos que, aunque encuentran mayores tasas de RC y de tiempo libre de enfermedad con tratamientos agresivos, no encuentran diferencias significativas en cuanto a la mortalidad final12,30,31.
La progresión en nuestros pacientes (20 %) es similar a la de otras series. El factor predictivo más importante de progresión fue la no RC al primer tratamiento, el cual fue una variable independiente en 4 modelos de regresión logística distintos. El único otro factor que se asoció a progresión fueron lesiones en mancha-placa (T1-T2) frente a eritrodermia-tumores (T3-T4); esto es lógico, puesto que estos últimos son estadios más avanzados y tienen menos camino para progresar. Aunque las diferencias no fueron estadísticamente significativas el 27 % de los pacientes T2 progresó a un estadio mayor frente al 17,4 % de los pacientes T1. Las diferencias no se asociaron a mayor progresión edad, sexo o retraso en el diagnóstico.
Casi dos tercios de los pacientes se encuentran vivos después de un tiempo medio de seguimiento de casi 8 años, la mitad de ellos libres de enfermedad y la otra mitad con micosis fungoide. El 17 % de los pacientes ha fallecido específicamente por linfoma y el resto de los fallecimientos han sido por causas diversas: tres de causa desconocida, dos carcinomas viscerales, dos patología pulmonar, un infarto de miocardio, una isquemia mesentérica, un coma hiperglucémico y uno por accidente de tráfico. La mortalidad por linfoma en el grupo T1 al diagnóstico fue de 5 %, mientras que para T2 fue de 20,8 %. Sin embargo, estas diferencias no fueron significativas. Esto mismo sucede en varios estudios, donde la mortalidad porcentualmente es superior en pacientes con estadio IB frente a IA, pero no siempre con significación estadística9,14. Sin embargo, otras series sí encuentran mayor mortalidad en pacientes T2 frente a T112,13; en estos dos trabajos además se compara la mortalidad con la de la población general, y se observa que no hay diferencias significativas con los pacientes T1. Además, Zackheim et al13 comparan la mortalidad en pacientes T2 subdividiéndola en manchas, con mortalidad similar a T1, y placas, con una mortalidad estadísticamente superior. Es posible que eso se deba a la profundidad del infiltrado, lo cual es acorde con los hallazgos de la serie de Marti et al15. Sorprendentemente, y al contrario que la mayoría de los trabajos la mortalidad por linfoma para el grupo T1 frente a T2-4 tampoco mostró diferencias significativas (p = 0,09), posiblemente arrastrado por la difícilmente explicable escasa mortalidad en los estadios IIB y III de nuestra serie.
No hubo diferencias en cuanto a mortalidad entre los pacientes sin adenopatías frente a los que sólo tenían adenopatías palpables; sin embargo, sí las hubo cuando se comparaban dichos pacientes con adenopatías afectadas histológicamente (p = 0,002). Es muy controvertido el papel pronóstico de las adenopatías palpables (sin afectación histológica) como factores predictivos de mortalidad o supervivencia; en nuestro estudio, como en muchas publicaciones6,12 no hay diferencias en la supervivencia a largo plazo, pero en un reciente trabajo, el grupo de Willemze14 encuentra peor pronóstico en los pacientes con adenopatías palpables con respecto a estadios IA-IB. En cualquier caso, la detección de adenopatías debería seguirse de su correspondiente estudio histológico siempre que fuera posible, pues hay concordancia en todos los trabajos de su papel predictivo como marcador de mal pronóstico cuando están afectadas histológicamente. Analizando el estadio TN (M no se analiza debido a que sólo hay 2 casos al diagnóstico) el estadio con mayor mortalidad por linfoma fue el IVA (66,6 %), lo cual fue estadísticamente significativo (p = 0,001). Llama la atención que no fallezca ningún paciente en estadio IIB, posiblemente por el bajo número de casos, aunque hay trabajos en los que los estadios tumorales sin afectación de adenopatías (como nuestros casos) tienen una supervivencia a los 5 años del 70-80 %11,13,14.
El análisis univariante mostró asociación de la mortalidad con la progresión de la micosis fungoide (p < 0,001), falta de remisión al primer tratamiento (p = 0,006), retraso comienzo-diagnóstico (p = 0,03), eosinofilia > 600 (p = 0,009), células de Sézary > 10 % (p = 0,0007), b2-microglobulina > 2,4 (p = 0,011), CD7 > 20 % (p = 0,001) y LDH > 300 (p = 0,004). Sin embargo, en el análisis multivariante se mantuvieron como variables pronósticas independientes la progresión de la enfermedad, la edad mayor a 60, el estadio N al diagnóstico (comparando N3 con N0-1) y la eosinofilia > 600. Con respecto a las pruebas de laboratorio, en la literatura se ha apuntado como factor de mal pronóstico las cifras elevadas de LDH9,15, β2-microglobulina9, y receptor soluble de IL-232; todos ellos probablemente indican un aumento de la masa tumoral en sangre periférica, de ahí su posible influencia en la evolución del proceso. En nuestro trabajo no se demostró asociación con las cifras de CD25 pero sí en el estudio univariante con LDH y β2-microglobulina; sin embargo, esa asociación se perdió en el estudio multivariante. Se han encontrado estudios de eosinofilia cutánea (en las lesiones de micosis fungoide) en las que ésta no influye de forma negativa15. Por otra parte, sólo se ha encontrado una referencia en la literatura en la que se implica como factor pronóstico negativo la eosinofilia en sangre periférica33. No obstante, se ha comunicado un perfil de linfocitos Th2 en síndrome de Sézary y en fases avanzadas de micosis fungoide34,35; el patrón de citocinas producido en estas fases podría explicar el aumento de eosinófilos y su valor como marcador de mal pronóstico. Sin embargo, serán necesarios más estudios para confirmarlo.
El análisis de supervivencia para la mortalidad por linfoma mostró, como es lógico, las mismas variables significativas descritas en el párrafo anterior con excepción de las células de Sézary circulantes superior al 10 % que se comportó como variable independiente de menor supervivencia en el estudio multivariante, mientras que en el análisis de la mortalidad sólo mostró asociación estadística en el estudio univariante. Diversos autores ya han comunicado este peor pronóstico en pacientes con linfocitos atípicos circulantes, además, según algunos es predictivo de afectación ganglionar y visceral6,11,36. Sigue creando controversia qué porcentaje o cuántas células de Sézary son necesarias para definir el síndrome de Sézary o influir en el pronóstico de los LCCT, puesto que pueden encontrarse en estadios muy iniciales de micosis fungoide e, incluso, en pacientes con dermatosis benignas. El Instituto Nacional de Cáncer Americano (NCI)18 usa el criterio de más del 5 %; no obstante, muchos centros suben ese porcentaje a 10 e incluso 20 % y no hay criterios unánimes. Nosotros hemos elegido de forma arbitraria un porcentaje del 10 %.
En nuestros pacientes las supervivencias fueron prolongadas, siguiendo más del 50 % de los pacientes vivos a los 23 años si se considera la mortalidad por linfoma, mientras que el 50 % han fallecido a los 18 años si se tiene en cuenta la mortalidad global. Estas supervivencias han oscilado entre 9 meses y 23 años desde el momento del diagnóstico y entre 13 meses y 49 años si tenemos en cuenta el comienzo de la enfermedad según los datos clínicos. En el momento actual, 61 pacientes han vivido 10 o más años desde el inicio de la enfermedad; de ellos 48 siguen vivos, 22 con ausencia de enfermedad (datos no mostrados). De los fallecidos, en ocho la causa ha sido un linfoma y en cinco otras causas.
Con el reordenamiento genético se ha realizado sólo estadística descriptiva (datos no mostrados) debido a la complejidad de la variable y el gran número de sesgos: en numerosos pacientes se ha determinado en varias ocasiones, en distintos momento de su enfermedad, en ocasiones con dos técnicas distintas (reacción en cadena de la polimerasa [PCR] y Southern-blot) con diferente sensibilidad y en ocasiones en diversos tipos de lesión cutánea (placa y tumor). Hay tres aspectos en este punto que merece la pena comentar. En primer lugar, la sensibilidad del reordenamiento por PCR fue algo superior respecto a Southern-blot, 56 % de las determinaciones frente a 49 % en piel y 36 % frente a 31 % en sangre periférica. En segundo lugar, reordenaron más las lesiones tumorales que las placas y reordenaron más los estadios TN más avanzados, tanto en piel como en sangre periférica. En tercer lugar, reordenaron en mayor porcentaje tanto los pacientes que progresaron como los que fallecieron por linfoma. Debido a los problemas mencionados se desconoce la significación de estos datos mediante un análisis multivariante; no obstante, parece que el reordenamiento positivo (en piel y sangre) puede ser una variable pronóstica en progresión y mortalidad por linfoma. Esto es congruente con un reciente trabajo que demuestra que la presencia de un clon en sangre periférica es un marcador pronóstico independiente37. Además, Delfau-Laure et al38 demostraron que la detección de un clon en piel era un factor predictor de respuesta al tratamiento, de tal modo que la posibilidad de obtener remisión completa en un paciente era mayor si no se detectaba ningún clon. Por último, un reciente trabajo muestra que el nivel PCR positivo o negativo (es decir, clonal o no) permanece constante en la gran mayoría de los casos, tanto en lesiones tomadas el mismo día como en muestras tomadas a lo largo del curso de la enfermedad38. En nuestro estudio esto no sucede, habiendo numerosos pacientes con reordenamientos positivos y negativos a lo largo de la evolución, tanto por PCR como por Southern. No obstante, son necesarios nuevos estudios para corroborar lo anterior.
CONCLUSIONES
Se han descrito las características clínicas y epidemiológicas de una serie de 100 pacientes. La mayoría de ellos son diagnosticados en estadios tempranos, con localización exclusivamente cutánea, cada vez con menos retraso desde el comienzo de la enfermedad hasta el diagnóstico final. No obstante, este retraso no influye en el pronóstico del paciente en cuanto a respuesta al tratamiento, progresión o mortalidad por linfoma.
Nuestro trabajo confirma los estudios previos en los que una proporción importante de pacientes con micosis fungoide, sobre todo en estadios iniciales, tienen buena respuesta al tratamiento inicial con RC duradera. Sólo una proporción, alrededor del 20 %, de estos pacientes fallece por causas directamente imputable a la micosis fungoide o progresa a estadios más avanzados de la enfermedad. La no RC al primer tratamiento es el marcador predictivo más importante de progresión tumoral.
En el estudio univariante fueron marcadores de mal pronóstico en cuanto a mortalidad/supervivencia la no RC al primer tratamiento, progresión a un estadio TNM mayor, adenopatías afectadas histológicamente, estadio IVA, edad > 60 años, células de Sézary circulantes > 10 %, CD7 > 20 %, eosinofilia > 600, β2-microglobulina > 2,4 y LDH > 600. A falta de estudio estadístico, la presencia de reordenamiento positivo en piel o sangre periférica constituye también un marcador de mal pronóstico. Se mantuvieron como variables independientes marcadoras de mal pronóstico en el estudio multivariante la progresión de la enfermedad, presencia de adenopatías afectadas histológicamente, células de Sézary circulantes > 10 %, eosinofilia mayor de 600 y edad de más de 60 años.
Son necesarios nuevos estudios para confirmar y conocer éstos y otros posibles factores de riesgo. Sería conveniente, en la medida de lo posible, una tipificación, no sólo en cuanto a diagnóstico, sino también en pruebas complementarias, seguimiento y tratamiento con vistas a unificar criterios y conseguir series que puedan ser comparables, para de este modo tratar de elucidar los factores de riesgo de progresión y mortalidad de estos procesos.


