




Epidermolysis bullosa (EB) is a heterogeneous group of inherited disorders characterized by a high degree of mucocutaneous fragility. This study aimed to describe the clinical and epidemiologic characteristics of patients with EB treated in Hospital Universitario La Paz, a national referral center for inherited EB.
Material and methodsObservational, retrospective, single-center study. We included all cases with a clinical and molecular diagnosis of EB managed in the hospital’s dermatology department from January 2, 2000, to February 28, 2021.
ResultsA total of 214 cases were studied. The median (interquartile range) age was 17 (8–32) years; 54.2% were women. One hundred thirty-five (63.1%) patients had dystrophic EB, 67 (31.3%) had EB simplex, 8 (3.7%) had junctional EB, and 3 (1.4%) had Kindler syndrome. One (0.5%) had EB acquisita. Over a third (35.5%) of the patients resided in Madrid. The most common clinical complications were pruritus (63.1%), local infections (56.5%), and pain (54.7%). The most serious ones were cardiomyopathy (in 5.6%) and squamous cell carcinoma (10.3%). Twenty-two patients (10.3%) died.
ConclusionsDystrophic EB was the most prevalent clinical form. The most prevalent complications were pruritus, pain, and infections. The most serious ones were cardiomyopathy and squamous cell carcinoma. This study is the first in Spain that explores strategies for improving the health status and quality of life of patients with EB.
La epidermólisis bullosa (EB) es un grupo heterogéneo de trastornos hereditarios caracterizado por un aumento de la fragilidad mucocutánea. El objetivo del presente estudio es describir las características clínicas y epidemiológicas de los pacientes con EB atendidos en el Hospital Universitario La Paz, centro de referencia nacional para EB hereditaria.
Material y métodoEstudio observacional, retrospectivo y unicéntrico. Se incluyeron todos los pacientes con diagnóstico clínico y molecular de EB atendidos en el servicio de Dermatología del Hospital Universitario La Paz desde el 1 de enero de 2000 hasta el 28 de febrero de 2021.
ResultadosSe registraron 214 pacientes, con una edad mediana de 17 años (RIQ: 8-32); el 54,2% fueron mujeres. Las formas clínicas correspondieron a EB distrófica con 135 (63,1%) casos, EB simple con 67 (31,3%) casos, EB juntural con 8 (3,7%), EB Kindler con 3 (1,4%) casos y EB adquirida con 1 (0,5%) caso. El 35,5% de los pacientes procedían de Madrid. Las complicaciones clínicas más frecuentes en nuestra serie fueron el prurito (63,1%), las infecciones locales (56,5%) y el dolor (54,7%). Las complicaciones más graves fueron las cardíacas (5,6%) y la aparición de CCE (10,3%). Fallecieron 22 pacientes (10,3%).
ConclusionesLa forma clínica predominante fue la EBDR. Las complicaciones más prevalentes fueron el prurito, el dolor y las infecciones, y las más graves, la miocardiopatía y el CCE. Es un estudio pionero realizado en nuestro país que permitirá́ implementar estrategias para mejorar la situación sociosanitaria de los pacientes con EB.
Epidermolysis bullosa (EB) is a heterogeneous group of inherited disorders characterized by increased mucocutaneous fragility and the formation of blisters, either spontaneously or after mechanical trauma.1 It is considered a rare disease, with an estimated prevalence of 6 cases/100 000 population in Europe.2
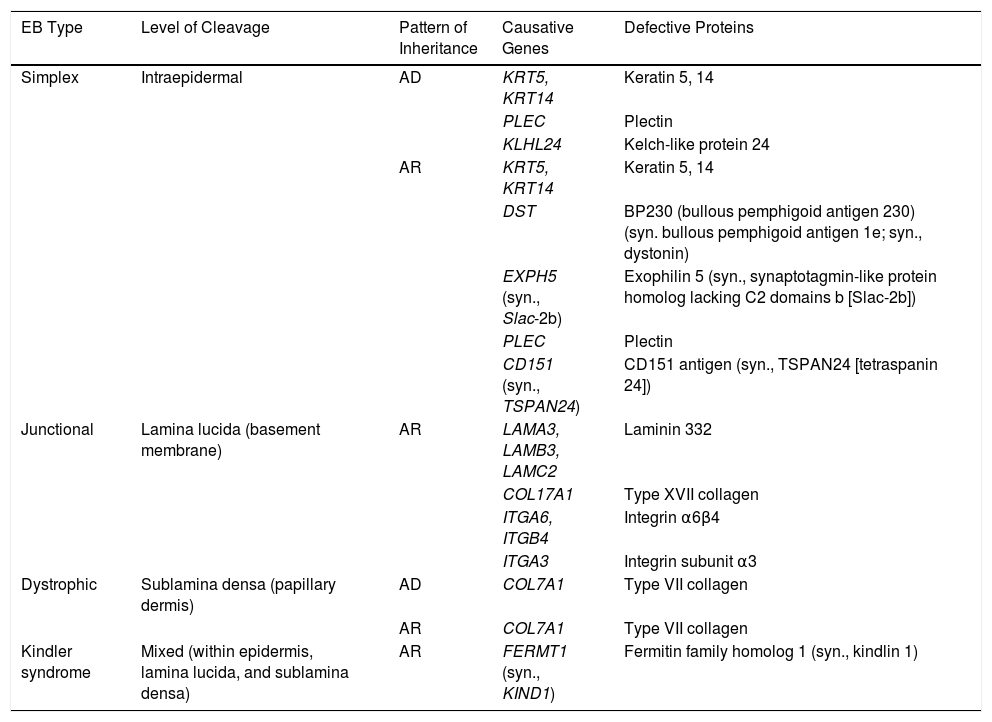
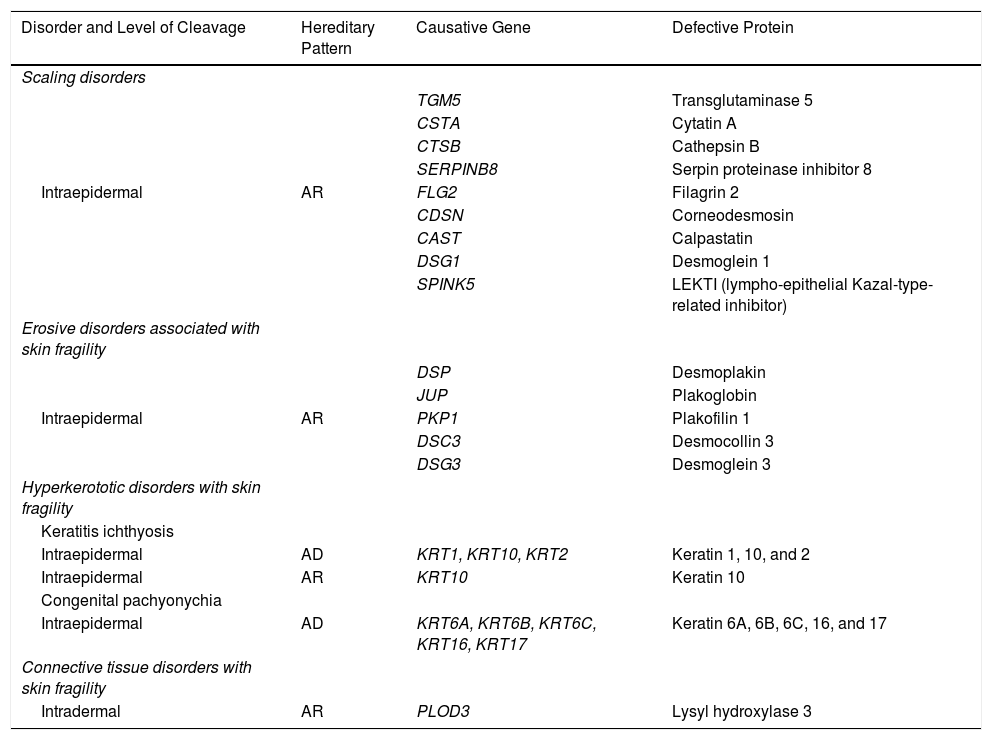
EB is caused by mutations in genes that code for proteins responsible for maintaining the integrity and mechanical stability of the integumentary system.3,4 Over 1000 mutations in 21 structural genes have been linked to skin adhesion defects and consequent fragility, giving rise to diverse subtypes of EB with cutaneous and extracutaneous abnormalities.1,5,6 Due to the large number of proteins implicated, diagnosis is complex and international classifications are revised periodically. More than 30 subtypes grouped in 2 categories are described in the latest classification.5,6 The first category encompasses classical forms of EB defined according to degree of skin blistering. It includes EB simplex, junctional EB, dystrophic EB, and Kindler syndrome (Table 1) (Fig. 1).The second category brings together other disorders with a tendency to cause blistering, but in which the main locus of disease and clinical manifestations are extracutaneous (Table 2). Supplementary Table S1 gives a summary of the specific features of classical EB subtypes.
Molecular Characteristics of the 4 Types of Classic EB.
| EB Type | Level of Cleavage | Pattern of Inheritance | Causative Genes | Defective Proteins |
|---|---|---|---|---|
| Simplex | Intraepidermal | AD | KRT5, KRT14 | Keratin 5, 14 |
| PLEC | Plectin | |||
| KLHL24 | Kelch-like protein 24 | |||
| AR | KRT5, KRT14 | Keratin 5, 14 | ||
| DST | BP230 (bullous pemphigoid antigen 230) (syn. bullous pemphigoid antigen 1e; syn., dystonin) | |||
| EXPH5 (syn., Slac-2b) | Exophilin 5 (syn., synaptotagmin-like protein homolog lacking C2 domains b [Slac-2b]) | |||
| PLEC | Plectin | |||
| CD151 (syn., TSPAN24) | CD151 antigen (syn., TSPAN24 [tetraspanin 24]) | |||
| Junctional | Lamina lucida (basement membrane) | AR | LAMA3, LAMB3, LAMC2 | Laminin 332 |
| COL17A1 | Type XVII collagen | |||
| ITGA6, ITGB4 | Integrin α6β4 | |||
| ITGA3 | Integrin subunit α3 | |||
| Dystrophic | Sublamina densa (papillary dermis) | AD | COL7A1 | Type VII collagen |
| AR | COL7A1 | Type VII collagen | ||
| Kindler syndrome | Mixed (within epidermis, lamina lucida, and sublamina densa) | AR | FERMT1 (syn., KIND1) | Fermitin family homolog 1 (syn., kindlin 1) |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; EB, epidermolysis bullosa; syn., synonym.

Molecular Characteristics of Genetic Disorders With Minor Skin Fragility.
| Disorder and Level of Cleavage | Hereditary Pattern | Causative Gene | Defective Protein |
|---|---|---|---|
| Scaling disorders | |||
| TGM5 | Transglutaminase 5 | ||
| CSTA | Cytatin A | ||
| CTSB | Cathepsin B | ||
| SERPINB8 | Serpin proteinase inhibitor 8 | ||
| Intraepidermal | AR | FLG2 | Filagrin 2 |
| CDSN | Corneodesmosin | ||
| CAST | Calpastatin | ||
| DSG1 | Desmoglein 1 | ||
| SPINK5 | LEKTI (lympho-epithelial Kazal-type-related inhibitor) | ||
| Erosive disorders associated with skin fragility | |||
| DSP | Desmoplakin | ||
| JUP | Plakoglobin | ||
| Intraepidermal | AR | PKP1 | Plakofilin 1 |
| DSC3 | Desmocollin 3 | ||
| DSG3 | Desmoglein 3 | ||
| Hyperkerototic disorders with skin fragility | |||
| Keratitis ichthyosis | |||
| Intraepidermal | AD | KRT1, KRT10, KRT2 | Keratin 1, 10, and 2 |
| Intraepidermal | AR | KRT10 | Keratin 10 |
| Congenital pachyonychia | |||
| Intraepidermal | AD | KRT6A, KRT6B, KRT6C, KRT16, KRT17 | Keratin 6A, 6B, 6C, 16, and 17 |
| Connective tissue disorders with skin fragility | |||
| Intradermal | AR | PLOD3 | Lysyl hydroxylase 3 |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive.
EB embraces a wide range of phenotypes, from the development of blisters on palms and soles to extracutaneous complications that can be life-threatening (Fig. 2). Epithelial tissues most at risk of complications are in the eyes, upper respiratory tract, esophagus, and urogenital area.7 Certain subtypes can cause serious complications in the musculoskeletal system, including the bone marrow, the heart, and the mouth. Such complications contribute to increased morbidity and mortality in EB. Potentially fatal squamous cell carcinomas (SCCs) can also appear in the second decade of life, mainly in patients with recessive dystrophic EB (RDEB).8

Extracutaneous complications in patients with RDEB. A, Patient with a local skin superinfection. B, Pseudosyndactyly on feet. C, SCC on the knee of a patient with RDEB. D, Fluorescein-positive ulcer on the corneal epithelium. RDEB refers to recessive dystrophic epidermolysis bullosa; SCC, squamous cell carcinoma.
In spite of great progress in our understanding of the molecular basis for the different EB subtypes, no cure for this disease has yet emerged. We have only preventive and symptomatic treatments for skin lesions and possible systemic complications.
Three hospitals were designated as reference centers for inherited EB in Spain in 2017. They are Hospital Universitario La Paz in Madrid and, in Catalonia, Hospital Sant Joan de Déu and Hospital Clínic de Barcelona. This study aimed to describe the clinical and epidemiologic characteristics of patients with EB attended in Hospital Universitario La Paz.
MethodsThis retrospective, observational single-center study carried out in Hospital Universitario La Paz included all patients with a clinical and molecular diagnosis of EB who were treated in the outpatient clinic of our hospital dermatology department’s specialist unit between January 1, 2000, and February 28, 2021.
Demographic data, molecular studies, and the EB subtype diagnosed were collected from records, along with mucocutaneous and extracutaneous complications observed during follow-up. The main qualitative descriptive variables were expressed as frequencies and proportions. The nonparametric Kaplan-Meier estimator was used to analyze survival. The log-rank test was used to compare overall versus subtype-associated mortality rates.
The study was approved by the ethics committee of Hospital Universitario La Paz. The committee considered that the project was limited to a secondary analysis of information the investigators had previously obtained during the course of clinical care.
ResultsA total of 214 patients were treated at the hospital during the study period. The median age was 17 years (interquartile range, 8–32 years); 54.2% were female. By subtype diagnosis, 135 had dystrophic EB (63.1%), 67 EB simplex (31.3%), 8 junctional EB (3.7%), 3 Kindler syndrome (1.4%), and 1 acquired EB (0.5%). Over a third (35.5%) of the patients were from Madrid. Patient characteristics are summarized in Table 3
Demographic Information and Diagnoses of the 214 Patients Treated for EB at Hospital Universitario La Paz Between 2000 and 2021.
| Characteristic | |
|---|---|
| Sex, n (%)a | |
| Female | 116 (54.2) |
| Male | 98 (45.8) |
| EB subtype, n (%)a | |
| Simplex | 67 (31.3) |
| Dystrophic | 135 (63.1) |
| Juntural | 8 (3.7) |
| Kindler syndrome | 3 (1.4) |
| Acquired | 1 (0.5) |
| Age, median (IQR) | 17 (8–32) |
| Spanish autonomous community of birth, n (%)a | |
| Madrid | 76 (35.5) |
| Castile-Leon | 27 (12.6) |
| Andalusia | 26 (12.1) |
| Castile-La Mancha | 18 (8.4) |
| Extremadura | 13 (6.1) |
| Community of Valencia | 12 (5.6) |
| Galicia | 11 (5.1) |
| Basque Country | 8 (3.7) |
| Canary Islands | 7 (3.3) |
| Aragon | 6 (2.8) |
| Catalonia | 2 (0.1) |
| Navarre | 2 (0.1) |
| Other | 6 (2.8) |
Abbreviations: EB, Epidermolysis bullosa; IQR, interquartile range.
The diagnosis was confirmed by genetic testing in 76.63%. The most common finding was a c.6527insC mutation in the COL7A1 gene, which was present in 21.5% of the patients overall and 34.1% of the patients with RDEB.
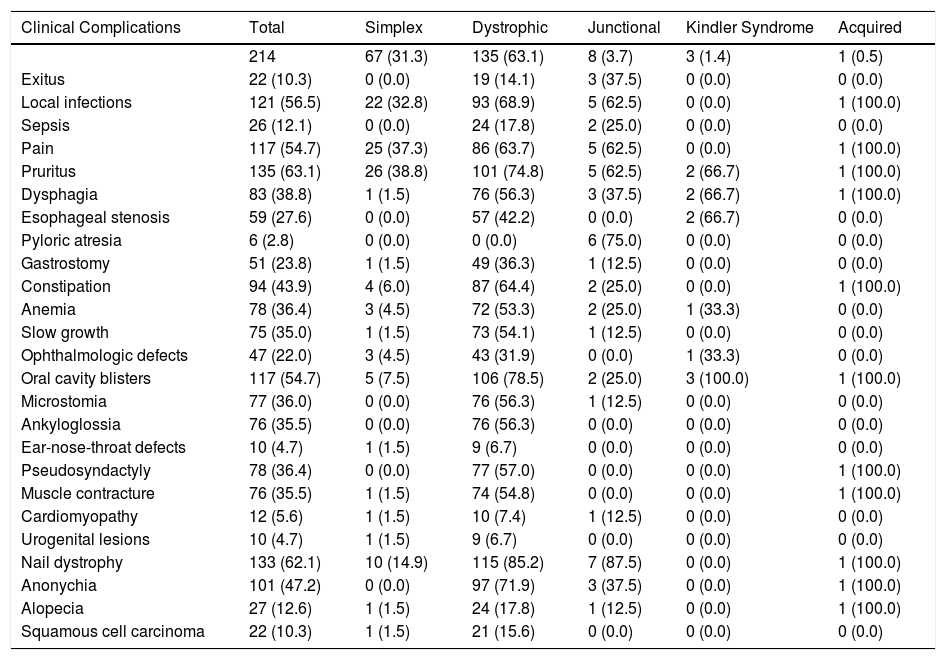
The main mucocutaneous and extracutaneous complications observed are listed in Table 4. The most common in all types of EB were pruritus (in 63.1%) and pain (in 54.7%). Local infections in open lesions were present in 56.5%; Pseudomonas aeruginosa and Staphylococcus aureus were most often implicated (in 45.22% and 41.44%, respectively). Twenty-six patients (12.1%) had severe sepsis, which led to the death of 2 patients with severe junctional EB. Forty-one percent of the patients required admission to the intensive care unit.
Clinical Complications of EB in Patients Treated in the Specialist Unit of Hospital Universitario La Paz Between 2000 and 2021, by Subtypes.a
| Clinical Complications | Total | Simplex | Dystrophic | Junctional | Kindler Syndrome | Acquired |
|---|---|---|---|---|---|---|
| 214 | 67 (31.3) | 135 (63.1) | 8 (3.7) | 3 (1.4) | 1 (0.5) | |
| Exitus | 22 (10.3) | 0 (0.0) | 19 (14.1) | 3 (37.5) | 0 (0.0) | 0 (0.0) |
| Local infections | 121 (56.5) | 22 (32.8) | 93 (68.9) | 5 (62.5) | 0 (0.0) | 1 (100.0) |
| Sepsis | 26 (12.1) | 0 (0.0) | 24 (17.8) | 2 (25.0) | 0 (0.0) | 0 (0.0) |
| Pain | 117 (54.7) | 25 (37.3) | 86 (63.7) | 5 (62.5) | 0 (0.0) | 1 (100.0) |
| Pruritus | 135 (63.1) | 26 (38.8) | 101 (74.8) | 5 (62.5) | 2 (66.7) | 1 (100.0) |
| Dysphagia | 83 (38.8) | 1 (1.5) | 76 (56.3) | 3 (37.5) | 2 (66.7) | 1 (100.0) |
| Esophageal stenosis | 59 (27.6) | 0 (0.0) | 57 (42.2) | 0 (0.0) | 2 (66.7) | 0 (0.0) |
| Pyloric atresia | 6 (2.8) | 0 (0.0) | 0 (0.0) | 6 (75.0) | 0 (0.0) | 0 (0.0) |
| Gastrostomy | 51 (23.8) | 1 (1.5) | 49 (36.3) | 1 (12.5) | 0 (0.0) | 0 (0.0) |
| Constipation | 94 (43.9) | 4 (6.0) | 87 (64.4) | 2 (25.0) | 0 (0.0) | 1 (100.0) |
| Anemia | 78 (36.4) | 3 (4.5) | 72 (53.3) | 2 (25.0) | 1 (33.3) | 0 (0.0) |
| Slow growth | 75 (35.0) | 1 (1.5) | 73 (54.1) | 1 (12.5) | 0 (0.0) | 0 (0.0) |
| Ophthalmologic defects | 47 (22.0) | 3 (4.5) | 43 (31.9) | 0 (0.0) | 1 (33.3) | 0 (0.0) |
| Oral cavity blisters | 117 (54.7) | 5 (7.5) | 106 (78.5) | 2 (25.0) | 3 (100.0) | 1 (100.0) |
| Microstomia | 77 (36.0) | 0 (0.0) | 76 (56.3) | 1 (12.5) | 0 (0.0) | 0 (0.0) |
| Ankyloglossia | 76 (35.5) | 0 (0.0) | 76 (56.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Ear-nose-throat defects | 10 (4.7) | 1 (1.5) | 9 (6.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Pseudosyndactyly | 78 (36.4) | 0 (0.0) | 77 (57.0) | 0 (0.0) | 0 (0.0) | 1 (100.0) |
| Muscle contracture | 76 (35.5) | 1 (1.5) | 74 (54.8) | 0 (0.0) | 0 (0.0) | 1 (100.0) |
| Cardiomyopathy | 12 (5.6) | 1 (1.5) | 10 (7.4) | 1 (12.5) | 0 (0.0) | 0 (0.0) |
| Urogenital lesions | 10 (4.7) | 1 (1.5) | 9 (6.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Nail dystrophy | 133 (62.1) | 10 (14.9) | 115 (85.2) | 7 (87.5) | 0 (0.0) | 1 (100.0) |
| Anonychia | 101 (47.2) | 0 (0.0) | 97 (71.9) | 3 (37.5) | 0 (0.0) | 1 (100.0) |
| Alopecia | 27 (12.6) | 1 (1.5) | 24 (17.8) | 1 (12.5) | 0 (0.0) | 1 (100.0) |
| Squamous cell carcinoma | 22 (10.3) | 1 (1.5) | 21 (15.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Abbreviation: EB: epidermolysis bullosa.
The oral mucosa was involved in some cases, in the form of ankyloglossia in 36% and microstomia in 35.5%.
Digestive system complications included dysphagia, found in 83 patients (38.8%) with severe dystrophic or junctional EB phenotypes. Esophageal stenosis was detected in 27.6%, all with RDEB. Nutrition was provided through a gastrostomy in 23.8% of the patients, most of whom had severe RDEB. The most common gastrointestinal symptom in all forms of EB was constipation, reported by 43.9% in this cohort. Pyloric atresia was found in only 6 patients (2.8%), all of whom had junctional EB. Slow growth was a characteristic of 75 patients (35%), most of them with RDEB. Seventy-eight patients (36.4%) had multifactorial anemia.
Musculoskeletal complications included muscle contractures in 76 patients (35.5%) and pseudosyndactyly in 78 (36.4%), in hands and/or feet. Ophthalmologic and urogenital complications occurred in 22% and 4.7%, respectively. Five patients had glomerulonephritis and 1 had meatal stenosis; all 6 of these patients had RDEB. Cardiomyopathy developed in 12 patients (5.6%), 10 with forms of RDEB, 1 with EB simplex, and 1 with junctional EB.
Twenty-two patients (10.3%) had SCC tumors. Thirteen of them (6.1%) had a single tumor and 9 (4.2%) had 2 or more; 21 of these patients had forms of RDEB, and 1 had EB simplex.
Twenty-two patients in this cohort (10.3%) died, due to severe sepsis (10 patients), end-stage renal failure (2), and aggressive metastatic SCC tumors (10). Survival analysis according to type of EB revealed significant differences (P = .011); dystrophic and junctional forms of EB (Fig. 3) were associated with higher mortality.
 in mortality were detected. The highest mortality was among patients with dystrophic or junctional EB. Although there were fewer deaths in the junctional EB group, survival time was shorter. Conversely, the dystrophic EB group had the larger number of deaths, survival time was longer than in the junctional EB group. EB refers to epidermolysis bullosa.")
Survival analysis by EB subtypes. Significant differences (P = .01) in mortality were detected. The highest mortality was among patients with dystrophic or junctional EB. Although there were fewer deaths in the junctional EB group, survival time was shorter. Conversely, the dystrophic EB group had the larger number of deaths, survival time was longer than in the junctional EB group. EB refers to epidermolysis bullosa.
Recurring mucocutaneous blisters and scarring are the main keys to identifying patients with EB. They are also related to most of the complications that develop.
Our observations in this cohort are comparable to reports published in other countries.7,8 Detailed knowledge of the frequency and clinical repercussions of EB complications, as well as of the subtypes with the most risk for them, are keys to early diagnosis and treatment.
The most prevalent EB subtype in our series was RDEB, with 135 patients (63.1%). This high percentage may be attributable to the fact that this subtype is associated with more clinical complications and therefore generates more patient referrals to specialists in the first years of life. EB simplex, which is the clinical form most often reported in other countries,2 appears to be underrepresented in our series. It may be that fewer EB simplex cases are in our records because of the absence of the severe complications that would lead them to our referral hospital.
The pathogenic c.6527insC mutation in the COL7A1 gene was the most common molecular finding in this cohort. This mutation was found in 34.1% of our patients with RDEB. This figure is somewhat lower than the one reported in an earlier population-based study of Spanish patients with EB, which found the mutation in 46.3% of the alleles of those with the RDEB subtype.9 This mutation, therefore, appears to be very common in Spain, inherited from some common ancestor; testing for the c.6527insC mutation is therefore a screening step in our patients with dystrophic EB.
The most common signs in all types of EB were pruritus (63.1%) and pain (54.7%). One hypothesis is that chronic skin lesions plus systemic inflammation leads to small fiber dysfunction and peripheral neuropathy, which would explain the pruritus and chronic pain.10 The highest prevalence of pruritus (74.8%) was seen in patients with dystrophic EB. Up to 93% of patients with this subtype have this symptom according to the literature; moreover it is known to be resistant to antihistamines, antiepileptics, and antidepressants.11,12
Infectious complications are common in all forms of EB. More than half the patients in our series had at least 1 episode of local superinfection, and such infections were most common in dystrophic EB, followed by junctional EB, and EB simplex. The usual culprit microorganisms were Paeruginosa and S aureus, as previously reported.13 In our cohort, these bacteria accounted for 45.22% and 41.44% of the infections, respectively. Sepsis developed in 12.1% and caused the death of 2 patients. This complication, which appeared in infancy almost exclusively in our patients with junctional EB, has been reported to be associated with a cumulative risk of between 11% and 20% at 1 year of age.14 While the risk is reported to be very low in other EB subtypes, it rises to about 8 percent by 35 years of age in RDEB.8
Oral mucosal involvement in the form of ankyloglossia and microstomia, appeared in 36% and 35.5% of our patients, respectively, and was seen more often in severe RDEB and less often in junctional EB, consistent with the literature.8,15 These manifestations reduce mouth size and make chewing and swallowing difficult, contributing to poor nutritional status.
We detected esophageal stenosis in 27.6% of our patients, all of whom had RDEB. Dysphagia was the main symptom. One study of 223 pediatric patients with EB reported a frequency of 64.9% for esophageal stenosis in RDEB,16 consistent with other series.7 This problem in patients with severe RDEB is common in infancy, and over half of these patients develop symptoms by the time they are 10 years old.16 The low prevalence in our series may be attributable to the fact that radiologic images are not ordered routinely and the lack of well-established protocols. Pyloric atresia, caused by mutations in the gene that codes for the α6β4 protein, develops early in forms of junctional EB,17 consistent with our finding of this complication in 2.8% of our patients.
Gastrointestinal symptoms were also present. The one most commonly associated with all forms of EB from the first years of life was constipation, present in 43.9% in our cohort. Constipation is estimated to occur in 20% to 40% of patients with EB simplex or junctional EB, and in 40% to 75% of those with RDEB,16,18 rates which are consistent with our findings.
Multifactorial anemia, which is common in severe forms of EB, developed in 36.4% of our patients; it is due to a combination of iron deficiency and chronic inflammation.19 Thirty-five percent of our patients showed delayed growth, consistent with findings reported for another cohort.16
Pseudosyndactyly in hands and feet and flexion contractures are frequent due to recurrent blistering and scarring.8 Pseudosyndactyly is typically seen more often in RDEB, as was the case in our cohort. One of the largest series published found that this complication developed after the first year of life in 13% to 16% of patients with severe RDEB and reached its highest prevalence (98.2%) in patients with severe RDEB at the age of 20 years.20
The most frequent ophthalmologic complications were corneal erosion and keratitis, which were observed in 22% in our cohort overall and in 31.9% of our patients with RDEB. These rates are lower than those reported in other series7,21 because such complications were not always properly recorded given that some patients were initially assessed in other Spanish autonomous communities.
The urogenital complications recorded, all in patients with RDEB, were glomerulonephritis (5 patients) and meatal stenosis (1 patient). Another study found a cumulative risk of mortality due to renal insufficiency to be 12.3% at age 35 years in patients with RDEB.22 Deaths among our patients with this form of EB occurred in the second decade of life, all due to kidney failure.
Respiratory-tract damage is observed mainly in severe cases of junctional EB.7 However, we saw this complication in only 1 of our patients, who had Dowling-Meara type EB simplex.
Heart defects were present in 5.6% of our patients. Dilated cardiomyopathy, a complication of severe RDEB, is rare but can be fatal, especially in patients who also develop chronic renal insufficiency. A high cumulative risk for this complication has been found for older patients with severe RDEB (4.5% after 20 years of age).23 However, cases at younger ages have also been reported in recent years according to a review of the literature,24 consistent with our cohort’s inclusion of 3 patients with severe dilated cardiomyopathy who were younger than 8 years old. This observation suggests the need for early echocardiography to screen for this complication in severe EB.
SCC is one of the main causes of death in patients with EB and occurs most often in RDEB, in which the cumulative risk of SCC ascends to 90.1% at 55 years of age according to one review.25 In our series the median age of detection of SCC was 26.5 years (interquartile range, 20–31 years), consistent with the literature.8 SCC is uncommon in EB simplex,26 yet 1 patient with this subtype in our cohort did develop this tumor. EB-associated SCC tumors are highly aggressive regardless of histopathologic features, and the recurrence and mortality rates are high.27
Twenty-two of our patients died, and the most frequent causes were sepsis and SCC. Severe junctional EB, with a life expectancy of less than 1 year,28 has the worst prognosis, as can be seen in the short survival time of the 3 patients with this diagnosis who died in our cohort.
The main limitations of this study are those intrinsic to retrospective analyses. It is possible that some complications were underrepresented in the records, so missing data may have affected the final figures. Furthermore, we did not look for evidence of psychological problems with specific assessment tools, even though high rates of anxiety and depression have been reported.29 Nor did we investigate the presence of skeletal complications, even though osteopenia and osteoporosis have been reported to be highly prevelant in severe EB.30
In conclusion, RDEB was the predominant form of EB in our practice. The most prevalent complications were pruritus, pain, and infections. The most serious ones were cardiomyopathy and SCC. The management of EB in specialized referral centers assures appropriate multidisciplinary assessment and follow-up under the leadership of dermatologists experienced with treating this disease.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
We thank the staff of our multidisciplinary team that treats patients with EB. We are also grateful for the unconditional support of the Spanish partner of the DEBRA Butterfly Children’s Charity (Asociación Piel de Mariposa DEBRA-España).
We thank the researchers of the following groups: CIBERER-CIEMAT-UC3M-IISFJD (Department of Bioengineering, Universidad Carlos III de Madrid); U714-CIBERER (Center for Biomedical Research of the Network for Rare Diseases); CIEMAT (Regenerative Medicine Unit); the Health Research Institute of the Jiménez Díaz Foundation; and the genetic diagnostic services for research into rare diseases of the Carlos III Health Institute (ISCIII).
Rocío Maseda Pedrero and Lucía Quintana Castanedo participated equally in writing the manuscript.
Please cite this article as: Maseda Pedrero R, Quintana Castanedo L, Pérez Conde I, Jiménez González M, Escámez Toledano MJ, de Lucas Laguna R. Epidermólisis bullosa en España: Estudio observacional de una cohorte de pacientes atendidos en un centro de referencia nacional, Actas Dermosifiliogr. 2021;112:781–793.


www.publicationethics.org.
