





La enfermedad de Fabry es la más frecuente por depósito de glucoesfingolípidos después de la enfermedad de Gaucher. Presenta una gran variabilidad clínica, siendo las manifestaciones en muchos de los órganos que se ven afectados inespecíficas1. Por este motivo, el diagnóstico de la enfermedad es difícil y suele retrasarse, como media unos 10 años después del inicio de la sintomatología2. La afectación cutánea en la enfermedad de Fabry es habitual y puede ser decisiva a la hora de sospechar el diagnóstico de la misma. Incluye la presencia de angioqueratomas, telangiectasias, alteraciones de la sudoración y linfedema. En trabajos recientes sobre las manifestaciones cutáneas en esta enfermedad se ha demostrado que el espectro clínico de los angioqueratomas es variado3. En este estudio, tras una revisión retrospectiva, se describen las lesiones cutáneas presentes en 5 pacientes diagnosticados de enfermedad de Fabry, prestando especial atención en la expresión clínica de los angioqueratomas.
Se trata de 5 pacientes, una mujer y 4 varones, afectados de una enfermedad de Fabry. Las principales características clínicas de cada uno de ellos se recogen en la tabla 1. La mayoría de ellos, incluida la única paciente mujer, presentaba una clínica extracutánea clásica. La edad media de aparición de los angioqueratomas fue de 17,2 años. En los casos 4 y 5, estos se distribuían de manera clásica, «en bañador», y en el caso 5 se observaron, además, unas lesiones vasculares menos hiperqueratósicas en ambas palmas. En los 3 casos restantes los angioqueratomas resultaron menos típicos. En el caso 1 se observaron lesiones aisladas en forma de mínimos angioqueratomas en la zona peribucal y periumbilical (fig. 1). En el caso 3, los angioqueratomas se distribuían de forma extensa, pero casi exclusivamente en el hemicuerpo izquierdo. Finalmente, la paciente heterocigota presentaba lesiones en forma de angioqueratomas en la hemivulva izquierda (fig. 2) y mínimas lesiones aisladas en la parte anterior del tronco. Únicamente en 2 casos había hipohidrosis como otra manifestación cutánea de la enfermedad. Todos los pacientes, a excepción del caso 1, recibían tratamiento enzimático sustitutivo (con una media de tratamiento ininterrumpido de 9 años) y en estos no se observó desaparición o disminución de los angioqueratomas, ni mejoría del resto de la clínica cutánea.
Hallazgos clínicos
| Caso | Sexo (H/M)/edad | Manifestaciones extracutáneas | Localización de los angioqueratomas | Edad de aparición angioqueratomas | Años de tratamiento enzimático | Otras manifestaciones dermatológicas |
| 1 | H/11 a | Intolerancia al ejercicioCórnea verticillata | Peribucal y alrededor ombligo | 7 a | No realiza tratamiento | No |
| 2 | M/36 a | AcroparestesiasIntolerancia al ejercicioTinnitusDiarreaCórnea verticillata | Región lumbar izquierda, vulva | 25 a | 9 a | Hipohidrosis |
| 3 | H/24 a | AcroparestesiasIntolerancia al ejercicioTinnitusDolor abdominalCornea verticillata | Hemicuerpo izquierdo | 6 a | 8 a | No |
| 4 | H/50 a | Hipertrofia del ventrículo izquierdoProteinuriaIntolerancia al ejercicioDiarrea | Flanco, cintura y cara interna de muslos | 34 a | 11 a | Hipohidrosis |
| 5 | H/32 a | AcroparestesiasTortuosidad vascular en ambos ojos | Muslos, nalgas y genitales, palmares | 14 a | 10 a | No |
a: años; H: hombre; M: Mujer.
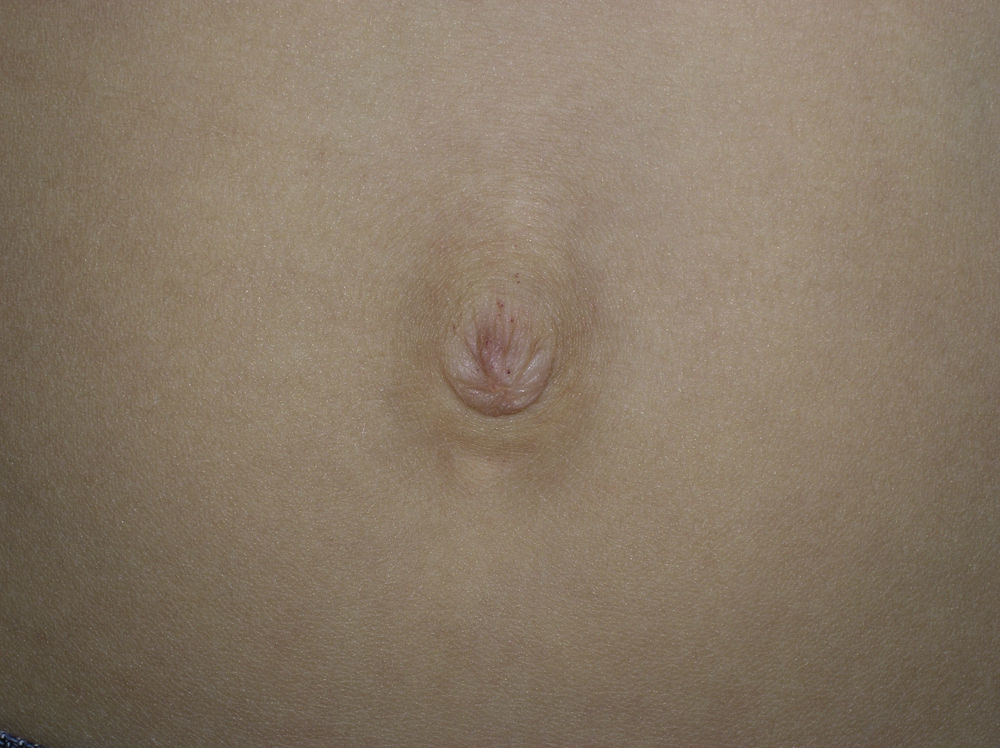

Dentro de los 5 subtipos de angioqueratomas que se reconocen4 se encuentra el denominado «angioqueratoma corporis difusum», muy característico de la enfermedad de Fabry aunque no exclusivo, ya que también puede verse en otras enfermedades por acumulación lisosómica5,6. Los angioqueratomas en la enfermedad de Fabry se encuentran en el 66% de los varones y el 36% de las mujeres, y pueden aparecer temprano en la infancia o bien en la vida adulta7. A pesar de que el término «angioqueratoma corporis difusum» pueda sugerir que los angioqueratomas en esta enfermedad son múltiples, no siempre es así, y como ocurrió en 3 de los 5 casos presentados, las lesiones pueden aparecer aisladas o agrupadas y pueden asentar en cualquier zona de la piel, las mucosas o los genitales. En las descripciones clásicas se destaca su distribución «en bañador», ya que con frecuencia asientan en la zona de los glúteos, los muslos y los genitales. En nuestra serie cabe destacar la distribución unilateral, afectando al hemicuerpo izquierdo de los angioqueratomas en un caso, y las lesiones mínimas limitadas al área peribucal, periumbilical y en la mucosa genital en otro de los pacientes. En esta última localización, pueden confundirse con los angioqueratomas de Fordyce.
La sospecha clínica de la enfermedad de Fabry, cuando las lesiones asientan en localizaciones menos típicas, es más difícil pero no debería pasar desapercibida. Aunque los síntomas propios de la enfermedad suelen iniciarse a partir de la primera década de la vida con acroparestesias, angioqueratomas, intolerancia al ejercicio, alteraciones oculares y gastrointestinales, las principales complicaciones se presentan en los adultos jóvenes cuando se desarrollan signos y síntomas renales, cardíacos y cerebrovasculares. Por tanto, y teniendo en cuenta que la enfermedad dispone de tratamiento específico que puede impedir el desarrollo de las complicaciones graves e irreversibles, es importante realizar un diagnóstico precoz que, en muchas ocasiones, podrá hacerse a partir de los angioqueratomas. En este sentido, el papel del dermatólogo puede ser fundamental.
El tratamiento enzimático sustitutivo (agalsidasa alfa) es el único tratamiento específico para la enfermedad y se ha demostrado que estabiliza la función renal y reduce la hipertrofia del ventrículo izquierdo8-10. En nuestra experiencia, el tratamiento sustitutivo no modificó ninguna de las manifestaciones cutáneas presentes antes de iniciarlo.
En conclusión, la observación de angioqueratomas, sea cual sea su localización y extensión, debe alertar al dermatólogo y obliga a poner en marcha las exploraciones complementarias pertinentes, ya que pueden permitir el diagnóstico de una enfermedad grave y potencialmente tratable. Asimismo, en los casos de nuevo diagnóstico, es importante estudiar a los familiares jóvenes para detectar una sintomatología que, en ocasiones, pasa desapercibida o se diagnostica de forma errónea.


