



Primary cutaneous diffuse cutaneous large cell lymphoma leg type (PCDCLCL-LT) accounts for approximately 3–4% of primary cutaneous lymphomas (PCL) and up to 20% of B-cell PCL. PCDCLCL-LT predominantly affects elderly patients and presents as aggressive lesions, usually on the lower extremities. However, up to 15–20% of cases may present in other sites, delaying diagnosis and complicating differential diagnosis.1
We conducted a retrospective analysis of nine cases diagnosed in 2 Spanish tertiary referral centers. We included all cases diagnosed with PCDCLCL-LT between 2016 and 2023, confirmed by histopathology and immunohistochemistry. No cases were excluded. Lesions were evaluated clinically, histologically, and immunophenotypically, and molecular testing was performed in 6 of the 9 cases. Molecular studies were conducted by next-generation sequencing (NGS) using a targeted 50-gene lymphoma panel. Fluorescence in situ hybridization (FISH) was performed in selected cases to detect rearrangements in MYC, BCL-2 and BCL-6.
The series (Table 1) included 9 patients (8 men and 1 woman), with a mean age of 79 years and a median follow-up of 9 months (range, 2–36 months). The median duration of lesions before diagnosis was 6 weeks (range, 2–14 weeks). Almost all patients had multiple lesions, with 7/9 (77.8%) being located on the legs. Two cases (22.2%) showed extracutaneous involvement. According to the International Society for Cutaneous Lymphomas/European Organization for Research and Treatment of Cancer (ISCL/EORTC) TNM classification, 3 patients were classified as stage T2b, 4 as stage T3a, and 2 as stage IV owing to extracutaneous involvement (cases 3 and 6). In patients with solitary or 2 localized lesions, local treatment with surgery or radiotherapy was prioritized. Specifically, patients #2 and #4 exhibited solitary lesions and were considered for local treatment: patient #4 underwent surgical excision, and patient #2 was not treated due to significant comorbidities and died shortly after diagnosis. Patient #5, who had 2 localized lesions and underwent complete surgical excision, remains in complete remission at the 14-month follow-up.
Characteristics of the sample.
| Case no. | Age | Sex | Elementary lesion | No. of lesions | Size of main lesion, cm | Location | Extracutaneous involvement | Molecular characteristics | Treatment | Evolution | Follow-up time (months) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 81 | M | Nodule | 5 | 3 | Left leg | No | MYC absent, BCL-2 and BCL6 not translocated | Mini R-CHOP X6 | CR | 36 |
| 2 | 89 | M | Tumor | 1 | 5 | Scalp | No | MYC absent, BCL-2 not translocated, BCL-6 translocated | Death 2 months after diagnosis (AMI) | Untreated | 2 |
| 3 | 88 | F | Nodule | 8 | 4 | Both legs | Hilar nodes, bone in leg | Non-translocated C-MYC, BCL-2 and BCL-6 | Mini R-CHOP X6 | Tumor progression and death by progression at 14 months | 14 |
| 4 | 64 | M | Nodule | 1 | 3 | Left leg | No | Non-translocated C-MYC, BCL-2 and BCL-6 | R-CHOP | Relapse at 3 months with subsequent CAR-T treatment and death after CAR-T (infection) at 12 months. | 12 |
| 5 | 70 | M | Tumor | 2 | 3 | Left leg, left arm | No | Non-translocated C-MYC, BCL-2 and BCL-6 | Surgery (excision) | CR | 14 |
| 6 | 75 | M | Tumor | 7 | 4 | Right leg | Bone in leg | Undetermined | R-CHOP | CR | 6 |
| 7 | 88 | M | Nodule | 1 | 4 | Right leg | No | Undetermined | Radiotherapy | Relapse after 1 month with subsequent CPR treatment and complete response | 5 |
| 8 | 73 | M | Plaque | Not applicable | Undetermined | Scrotum, hypogastrium | No | Undetermined | R-CHOP | Relapse after 2 months, with retreatment with R-CHOP and complete response | 11 |
| 9 | 82 | M | Nodule | >10 | 5 | Right leg | No | Undetermined | R-CP | CR | 9 |
AMI: acute myocardial infarction; BCL-2: B-cell lymphoma 2 gene; BCL-6: B-cell lymphoma 6 gene; CAR-T: chimeric antigen receptor T-cell therapy; CR: complete response; C-MYC: MYC oncogene; F: female; M: male; Mini R-CHOP: mini rituximab, cyclophosphamide, adriamycin, vincristine, prednisone; R-CHOP: rituximab, cyclophosphamide, doxorubicin, vinblastine, prednisone; R-CP: rituximab, cyclophosphamide, prednisone.
In all cases, histopathological examination showed a diffuse and monotonous proliferation of centroblastic and immunoblastic cells. The immunophenotypic profile was consistent: positivity for CD20, PAX5, BCL-2, BCL-6 and MUM1 and negativity for CD5, CD10 and CD30. The proliferative index measured by Ki-67 was above 75% in all cases. Molecular analysis identified some common alterations such as rearrangements in BCL2 and BCL6 and, to a lesser extent, c-MYC. Patient #5 showed MYD88 L265P, NFKBIE p.Y28fs, and a loss-of-function mutation in ARID1B. In addition, a monoclonal rearrangement in the immunoglobulin heavy chain was found in skin biopsies in cases #1, #2, #3, #4 and #6. No other significant molecular alterations were found in the remaining patients who underwent molecular testing.
The treatment of choice was polychemotherapy with R-CHOP or mini-R-CHOP in most patients, which is consistent with current recommendations for aggressive B-cell lymphomas. However, in cases with localized disease, particularly solitary lesions, surgical excision or radiotherapy was considered as first-line therapy. For patients with relapsed or refractory disease, management was discussed in a multidisciplinary tumor board involving dermatology and hematology, and treatment was individualized based on clinical status, comorbidities, and prior therapies. Patient #5, who harbored MYD88, NFKBIE and ARID1B mutations, underwent the surgical excision of 2 localized lesions and remained in complete remission. Three patients died during follow-up: 1 due to disease progression (patient 3), 1 after second-line CAR-T therapy (patient 4), and 1 from an acute myocardial infarction unrelated to lymphoma (patient 2). Among the 6 surviving patients, all were alive without clinical or radiologic evidence of disease at the last follow-up and did not require additional treatment.
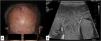
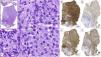
In our series, we wanted to highlight patient #2 (Fig. 1A) and patient #8 (Figs. 1B and 2) due to their presentation in atypical locations. Patient #2 presented with a tumor at the vertex and patient #8 with lesions at genital level. Specifically, the latter was diagnosed based on the presence of a massive edema in the scrotum occupying a large part of the subcutaneous cellular tissue (SCT). Despite an initial diagnosis of scrotal cellulitis (Fig. 1B), the patient's poor clinical evolution prompted an incisional biopsy (Fig. 2), which revealed diffuse infiltration of the dermis and subcutaneous tissue by a neoplasm composed of highly atypical B-cell lymphoid cells of medium to large size. The cells exhibited marked nuclear pleomorphism, abundant karyorrhexis, and a diffuse growth pattern arranged in loose, trabecular, and sheet-like formations.


Histopathology of patient number 8. (A) Diffuse lymphocytic proliferation of sheet-like growth with the presence of centroblasts (green arrow) and immunoblasts (red arrow). (B–D) Neoplastic cellularity was positive for CD20, BCL-2 and MUM1, among others. (E) A high proliferative index (Ki-67) was observed.
PCDCLCL-LT carries a guarded prognosis because of its high aggressiveness, particularly in older patients. However, rare atypical cases of spontaneous remission have been reported in the literature.2 In our series, most patients had a favorable outcome.
Although the most common site is the legs, atypical sites such as the scrotum or scalp in our series, although rare, highlight the need to include G-CSCL-PT in the differential diagnosis of lymphoproliferative lesions with secondary skin involvement. In patient 8, although the testes were free of neoplastic infiltration, this case underscores the similarities between leg-type lymphoma and secondary cutaneous involvement of diffuse large B-cell lymphoma, which may be histopathologically and immunophenotypically indistinguishable from PCDCLCL-LT.3
Accurate identification of PCDCLCL-LT by histological and immunophenotypic techniques is critical for proper management: neoplastic cells characteristically express BCL2 and MUM1. Though less frequent, positivity for BCL6 (and rarely for CD10) indicates a germinal center origin. In addition, a Ki67 positivity of more than 70% allows us to exclude most inflammatory dermatoses and helps us to assess anisokaryosis due to its characteristic nuclear staining.
Although MYD88 L265P mutations have been associated with aggressive clinical behavior in B-cell lymphomas,4–7 patient #5 from our series, who harbored this mutation, achieved complete remission with surgery alone. This finding suggests that the presence of MYD88 mutations may not invariably indicate poor prognosis in PCDCLCL-LT and underscores the importance of interpreting molecular alterations in the broader context of clinical presentation, histopathological features and anatomical site when assessing prognosis and individualizing treatment strategies.
Our findings are consistent with the largest published series to date, conducted by Grange et al. in France, which analyzed a total of 60 cases of PCDCLCL-LT in a multicenter setting. In their study, the 5-year disease-specific survival rate was 41%, and both leg involvement and multiple skin lesions at diagnosis were identified as independent adverse prognostic factors.8 Although our series includes fewer patients, the estimated 5-year disease-specific survival rate was approximately 55%. We also observed that multiple lesions and extracutaneous involvement were associated with worse outcomes, while solitary or atypically located lesions (patients #2 and #8) did not confer a poor prognosis. To our knowledge, this is one of the largest Spanish series specifically focused on PCDLBCL-LT, and complements existing literature by offering a contemporary clinicopathological and molecular characterization of this rare lymphoma subtype.
In conclusion, this case series highlights 2 main contributions: the presence of atypical anatomical presentations, and the molecular characterization of a MYD88-mutated case with favorable outcome. These findings underscore the importance of a multidisciplinary approach and individualized assessment based on clinical, histopathological and molecular features.
Conflict of interestThe authors declare that they have no conflict of interest.


