


El dermatofibrosarcoma protuberans (DFSP) es un tumor cutáneo, localmente agresivo, que afecta a adultos jóvenes o de edad media. Se han descrito diferentes formas histológicas, siendo la mixoide una de las más infrecuentes. Microscópicamente está constituido por una neoformación que ocupa la dermis e hipodermis, de predominio mixoide, con áreas periféricas conformadas por haces de células fusiformes, de crecimiento infiltrativo, que expresan el marcador inmunohistoquímico CD34 de forma difusa. Presentamos por primera vez un DFSP mixoide en un dedo de la mano izquierda de una niña de 14 años, de más de 10 años de evolución. Se trata del primer caso infantil de DFSP mixoide en dicha localización. Este subtipo histológico se ha descrito fundamentalmente en las extremidades de adultos, siendo excepcional en niños. Se comenta el diagnóstico diferencial con otros tumores mesenquimales mixoides CD34 positivos.
Dermatofibrosarcoma protuberans is a locally aggressive skin tumor that affects young and middle-aged adults. A number of histological variants have been described, the myxoid type being one of the least common. Microscopically it is formed of a neoplastic growth that is located in the dermis and hypodermis and has a predominant myxoid component. Peripherally there are infiltrating bundles of spindle-shaped cells that are diffusely positive for the CD34 immunohistochemical marker. We report a case of myxoid dermatofibrosarcoma protuberans on a finger of the left hand of a 14-year-old girl. The tumor had been present for at least 10 years. This is the first pediatric case of myxoid dermatofibrosarcoma protuberans at this site. This histological subtype has mainly been described on the extremities in adults and is very rare in children. We discuss the differential diagnosis with other CD34+ myxoid mesenchymal tumors.
El dermatofibrosarcoma protuberans (DFSP) en un tumor mesenquimal con diferenciación fibroblástica y miofibroblástica localizado en la dermis y tejido celular subcutáneo de adultos jóvenes o de edad media1,2, con un pico de incidencia en la cuarta década de la vida3. Es un tumor de crecimiento lento y malignidad intermedia, localmente agresivo, que recidiva en más de un tercio de los casos y metastatiza solo de forma ocasional, preferentemente por vía hematógena. Un 10-15% de los casos recidivantes desarrollan áreas de fibrosarcoma, neurofibrosarcoma o mixofibrosarcoma3–5.
Se han descrito diversos tipos de DFSP entre los que destacan: el clásico, pigmentado (tumor de Bernard), fibrosarcomatoso, de células granulares, con diferenciación miogénica, plano atrófico y el mixoide1,2. El subtipo fibrosarcomatoso implica un peor pronóstico, con un riesgo de metástasis del 10-15% y de muerte relacionada del 5,8%6,7. Una de las variantes más infrecuentes es la mixoide, de presentación en extremidades, cabeza, cuello y tronco de adultos, siendo muy rara en niños, en los que solo se ha descrito en 3 ocasiones3. Presentamos por primera vez un caso de DFSP mixoide en el dedo de la mano de una niña de 14 años.
Caso clínicoPaciente de 14 años, que acudió al Servicio de Dermatología de nuestro centro por presentar una tumoración lobulada, de 2,5cm de diámetro mayor, en la superficie dorsal de la falange media del tercer dedo de la mano izquierda (fig. 1). La paciente refería que la lesión llevaba más de 10 años de evolución, era de crecimiento lento y progresivo, y que había presentado dolor en los últimos meses. No se logró obtener el dato de si la lesión estaba presente desde el nacimiento. Al examen físico, la lesión era de consistencia variable, alcanzaba planos profundos, no se desplazaba con los tendones y no limitaba el movimiento del dedo.
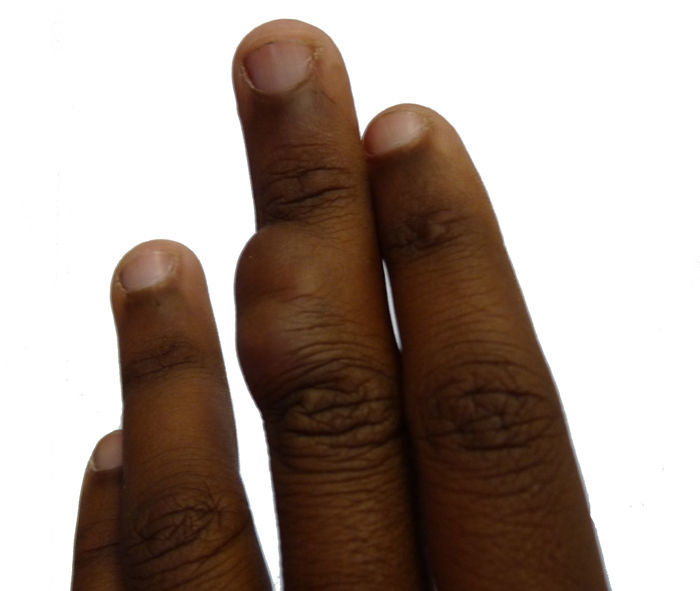
Se procedió a la toma inicialmente de una biopsia incisional y tras el diagnóstico histopatológico, se decidió extirpar completamente la lesión. Se realizó una extirpación quirúrgica simple con aproximación de bordes, hasta la fascia tendinosa, buscando márgenes laterales superiores a 1cm con respecto a la lesión clínica. En el acto quirúrgico, se apreció que el tumor era de consistencia variable, con zonas blandas de aspecto mixoide, sin infiltración de los tendones subyacentes. Seis meses después de la exéresis la paciente no ha presentado recidiva.
Estudio histopatológicoTanto el material de la biopsia incisional como el de la extirpación quirúrgica se fijaron en formalina tamponada al 10% y se incluyeron en su totalidad de forma rutinaria en parafina. Se realizaron cortes de 3 micras de espesor que se tiñeron con hematoxilina-eosina. Posteriormente, se efectuó estudio inmunohistoquímico con los anticuerpos CD34 (DAKO, 1:50), proteína S-100 (DAKO, 1:5000), factor XIIIa (Behring, 1:1000), desmina (DAKO, 1:100), actina 1A4 (DAKO, 1:50), Ki-67 (DAKO, 1:300), EMA (DAKO, 1:100) y bcl-2 (DAKO, 1:80), mediante el método ABC, con controles positivo y negativo. Se realizó hibridación in situ con fluorescencia (FISH) de tejido tumoral embebido en parafina para detección de la translocación t(17;22)(q22;q13).
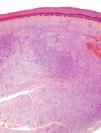
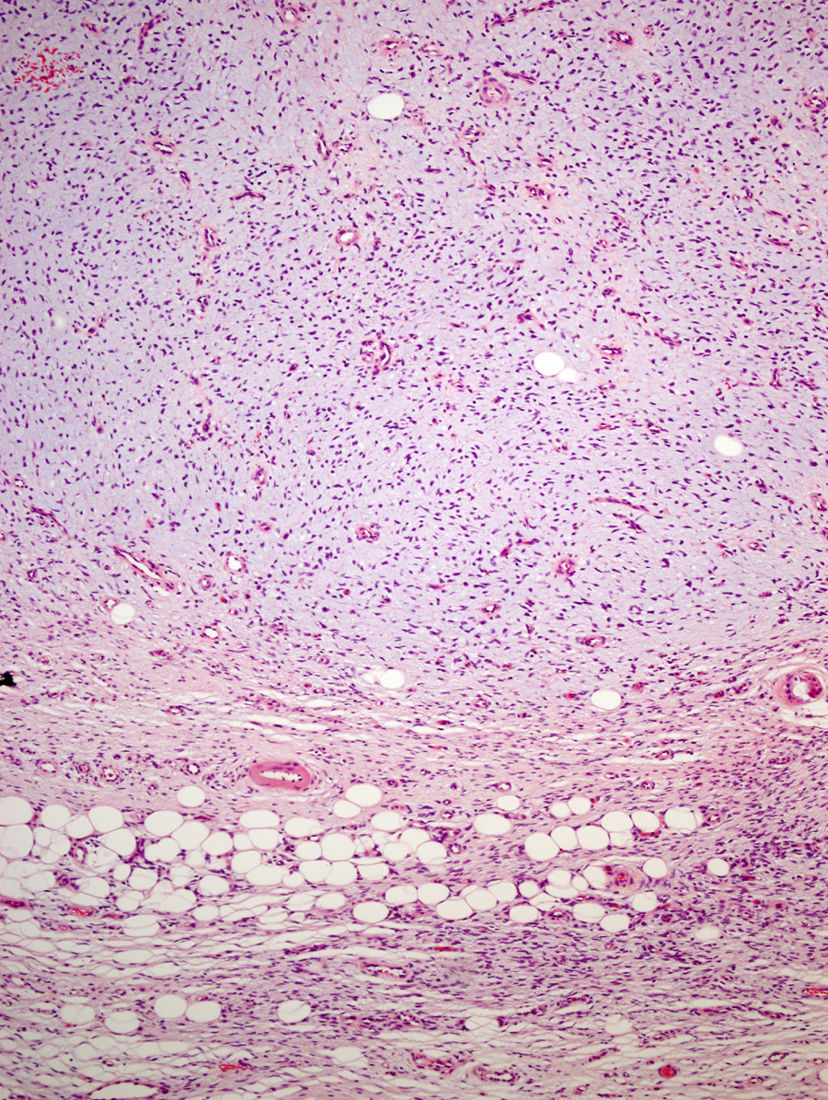
La biopsia incisional presentaba una ocupación difusa de la dermis reticular por una neoformación conformada por haces cortos de células fusiformes, que se entrecruzaban en diferentes direcciones, dejando una zona de Grenz respetada. El tumor rodeaba los anejos cutáneos sin invadirlos ni destruirlos (fig. 2). Las células proliferantes poseían un citoplasma eosinofílico de límites imprecisos y núcleos alargados, ovales, de cromatina fina, sin nucleolo. Solo se identificaron dos figuras de mitosis, no atípicas, por 10 campos de gran aumento. En la porción profunda, la lesión era menos celular y poseía áreas estromales de aspecto mixoide.
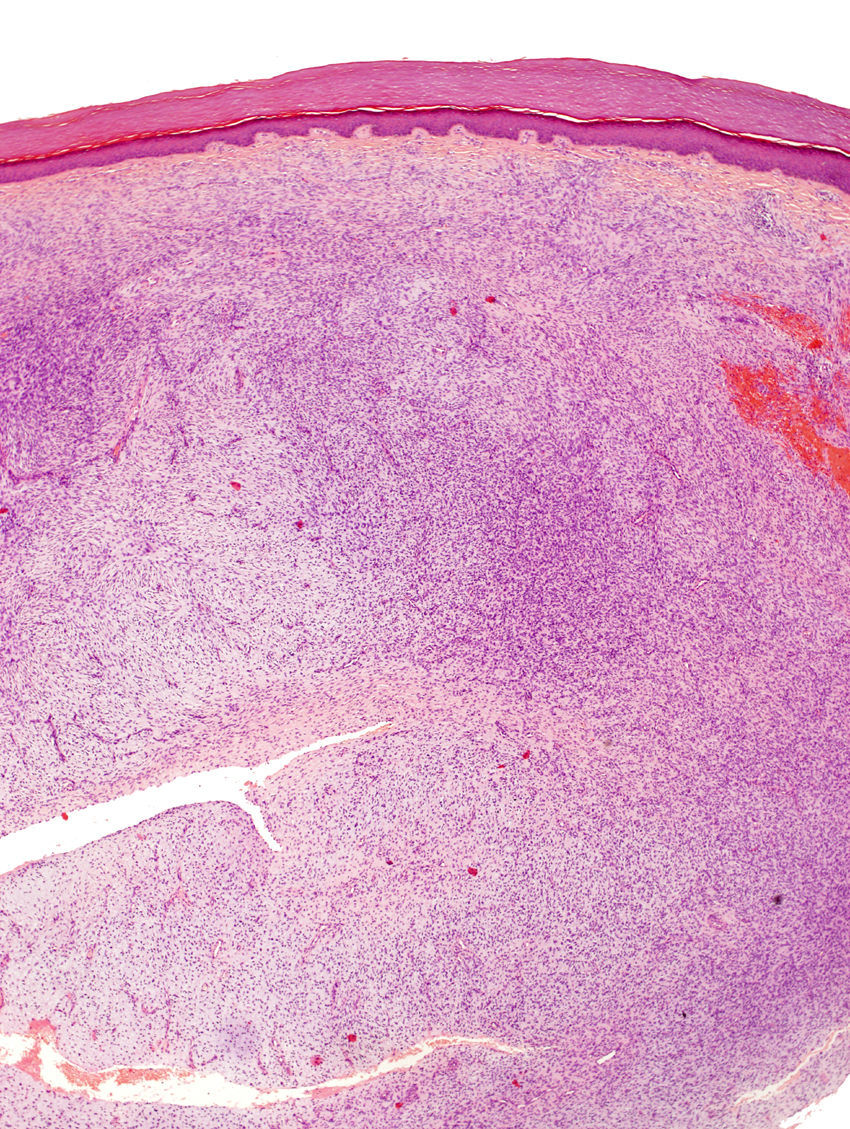
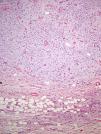
La pieza de extirpación consistía en una elipse de piel en la que se identificaba un nódulo de 2,4cm de eje máximo. Histológicamente estaba constituido por una neoformación mesenquimal de patrón de crecimiento difuso, de bordes infiltrativos, que ocupaba toda la dermis reticular y que se extendía al tejido celular subcutáneo, con un patrón en «panal de abeja», aislando en forma individual a los adipocitos (fig. 3). Sin embargo, un 90% de la neoformación estaba conformada por zonas hipocelulares de aspecto mixoide, en las que las células eran fusiformes y estrelladas, sin atipia ni mitosis, con numerosos vasos capilares de paredes finas y escasas células inflamatorias. Focalmente se observaba un estroma denso fibroso. En la periferia, tanto en la porción superficial como en la profunda, se observaban áreas focales con las mismas características descritas en la biopsia incisional. La lesión afectaba de forma amplia los márgenes quirúrgicos de resección laterales y el profundo.
La tinción de hierro coloidal fue positiva (fig. 4) y con las técnicas de inmunohistoquímica, se observó intensa expresión citoplásmica para CD34 en las áreas de patrón clásico y débil en las áreas mixoides. El resto de marcadores realizados fueron: proteína S-100, factor XIIIa, actina 1A4, desmina, EMA y bcl-2 resultaron negativos. El índice de proliferación celular con Ki-67 fue de un 5% en las zonas clásicas y de un 20% en las áreas mixoides. El diagnóstico de DFSP fue confirmado demostrando la translocación t(17;22)(q22;q13) mediante FISH.
Discusión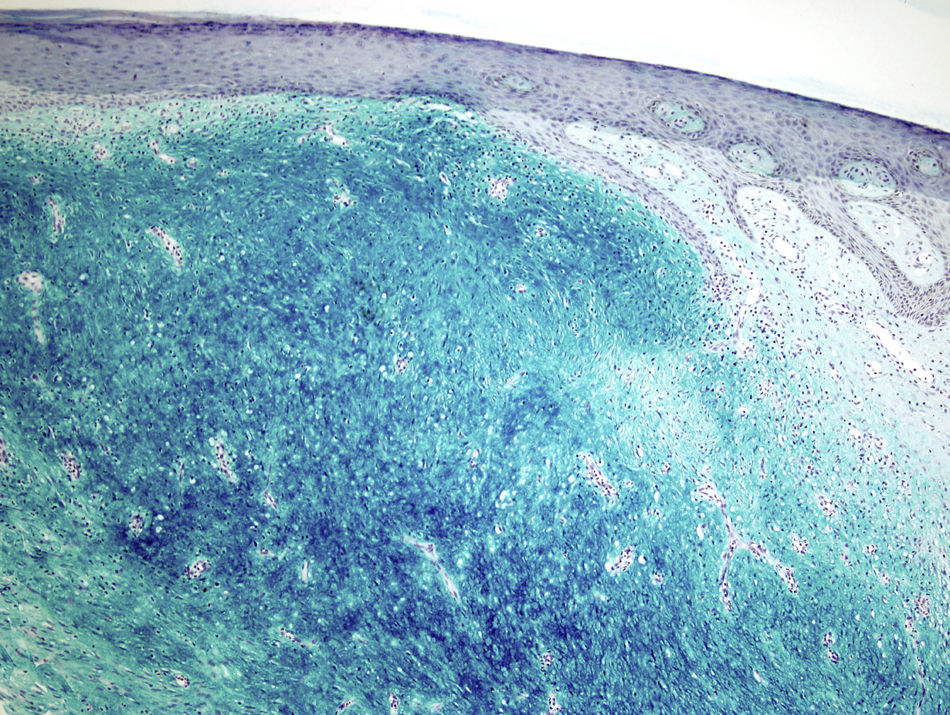
El subtipo mixoide es una variedad muy infrecuente de DFSP, descrita por primera vez en 1983 por Frierson et al9. En una de las series más grandes publicadas de DFSP, solo un 4,3% correspondió a la variante mixoide3. La mayoría de los casos, a diferencia de la variedad clásica, se presentan en las extremidades, seguidos en frecuencia por la cabeza y el cuello, el tronco y la región anogenital3. El DFSP mixoide es un tumor muy raro en niños. Solo se han descrito 3 casos en la infancia, localizados en párpado, cuero cabelludo y codo, todos en pacientes de sexo masculino3,4. Nuestro caso representa el primer DFSP mixoide en el dedo de una niña.
Clínicamente, se presenta como una masa nodular dérmica o subcutánea, firme, elevada, de crecimiento lento y progresivo3. Los diagnósticos clínicos presuntivos más frecuentes son: quiste epidérmico, mucinoso o sinovial, lipoma, granuloma piogénico o linfoma3. Nuestro caso fue remitido, debido a su localización, como tumor de células gigantes de la vaina tendinosa.
En relación a la evolución, algunos autores refieren que la variante mixoide posee un pronóstico levemente mejor que la clásica, mientras que otros no1,3. En una serie reciente de 23 casos de DFSP mixoide, no se observó metástasis en ninguno de ellos. En la misma serie, dos pacientes presentaban recidiva local, uno con resección incompleta y el otro con márgenes inferiores a 2mm3. Tampoco se han descrito metástasis en niños4. Nuestro caso presentó un crecimiento lento y progresivo durante más de 10 años, sin recidivas ni metástasis 6 meses después de la exéresis.
Microscópicamente, el tumor se localiza en la dermis reticular y tejido celular subcutáneo, dejando una zona de Grenz respetada, aunque ocasionalmente afecta la epidermis y la úlcera5. Predominan las áreas con estroma mixoide, basofílico o pálido, debido al alto contenido de ácido hialurónico, evidenciado con la tinción de hierro coloidal. Es probable que la transformación mixoide se deba a un aumento en la síntesis de glicosaminoglucanos, lo que inhibe la polimerización del colágeno8. No es infrecuente que el DFSP clásico tenga áreas focales de aspecto mixoide, sobre todo en las recidivas, sin que esto implique que se trate de la variante mixoide. Este subtipo se diagnostica cuando más del 50% del estroma de la lesión es mixoide3,8. En el presente caso, el 70% del tumor era mixoide. El estroma mixoide contiene escasas células no atípicas, fusiformes y estrelladas, de citoplasma levemente eosinofílico o pálido, de bordes no definidos, con núcleos ovales, de cromatina fina, sin nucleolo y ocasionales o nulas mitosis1,5. Además, el tumor contiene capilares de paredes finas distribuidos al azar, que raramente se acompañan de mastocitos. Pueden identificarse también células dendríticas pigmentadas como en el tumor de Bernard3,5,9. Una de las claves del diagnóstico es la presencia, en la periferia de la lesión, de áreas características de DFSP clásico, conformadas por células fusiformes, con una disposición en haces que se entrecruzan, con escaso colágeno entre ellos y áreas en «rueda de carro». La lesión rodea los anejos cutáneos sin invadirlos y cuando infiltra el tejido adiposo subcutáneo adopta un patrón característico en «panal de abeja», al rodear en forma individual a los adipocitos1,3,5,8,9. En nuestro caso, un 30% del tumor correspondía a la variante clásica y presentaba en la periferia áreas de infiltración de la grasa subyacente.
El marcador inmunohistoquímico más específico es el CD34, con positividad difusa e intensa en el 50-100% de los casos y una sensibilidad del 84-100%. En las áreas mixoides y fibrosarcomatosas, la tinción es débil, focal o negativa3,5,6. Nuestro caso presentaba positividad difusa con mayor intensidad en las escasas áreas celulares. Además el tumor es positivo para vimentina y CD99. Algunos casos son focalmente positivos con el factor XIIIa5. Puede haber expresión de EMA en las áreas mixoides, lo que podría reflejar diferenciación perineural10, pero no expresa S-100, actina ni desmina5. Se han reportado índices de proliferación celular (Ki-67) mayores en las áreas mixoides (19,8%) y fibrosarcomatosas (11,8%) que en las zonas clásicas (2,2%)11. De hecho, en nuestro caso el índice proliferativo (Ki-67) en las áreas mixoides era de un 20%, bajando hasta un 5% en las áreas más densamente celulares.
El DFSP se caracteriza por estar asociado a la translocación recíproca t(17;22)(q22;q13) o a un cromosoma supernumerario en anillo r(17;22). Ambas resultan en un gen quimérico COL1A1-PDGFB, por la fusión del gen del colágeno tipo I alfa del cromosoma 17 y el gen de la cadena beta del factor de crecimiento derivado de las plaquetas del cromosoma 22. Esto provoca una estimulación continua del crecimiento celular1,6. La translocación se describe con mayor frecuencia en niños, mientras que en adultos predominan los anillos cromosómicos, lo que podría representar un evento tardío en la patogénesis del DFSP4. Esta alteración genética se ha encontrado también en la variante mixoide y en el fibroblastoma de células gigantes5. Actualmente, el tratamiento con inhibidores de tirosina kinasa selectivos para PDGFR se reserva para casos de enfermedad irresecable, recidivante o metastásica6. La inmunorreactividad positiva con el anticuerpo PDGFR-B indica activación constitucional del receptor PDGF, lo que constituye un método indirecto alternativo, para confirmar la presencia no regulada del gen PDGF afectado con la translocación6.
Ante una lesión cutánea/subcutánea con predominio de áreas mixoides y con positividad para CD34, el patólogo debe plantearse el diagnóstico diferencial con lesiones mesenquimales benignas y malignas, de comportamientos biológicos y conducta terapéutica diferente1, especialmente si la muestra es superficial y pequeña. La presencia de zonas clásicas de DFSP, aunque focales, suelen ser de gran ayuda diagnóstica3. En el presente caso, la biopsia incisional mostraba zonas clásicas de DFSP, lo que facilitó el diagnóstico inicial, que hubo de replantearse al estudiar el espécimen de la excisión, de predominio mixoide.
El principal diagnóstico diferencial en este caso debe hacerse con el fibromixoma acral superficial (FAS), que suele afectar a pacientes de entre 14 y 72 años de edad, crece como una masa solitaria de 0,6 a 5cm de diámetro, con una evolución de 3 meses a 30 años y con predilección por los dedos de las manos y los pies, con tendencia a afectar la uña. Las recurrencias son frecuentes después de resecciones parciales12. Ambos tumores se localizan en la dermis y tejido subcutáneo, están constituidos por células estrelladas y fusiformes con un patrón de crecimiento fascicular, al azar o en «rueda de carro», en una matriz mixoide o colagenosa, con vascularización moderada, atipia leve y escasas mitosis. Además ambos tumores expresan CD34, EMA y CD99 y son negativos para S-100, actina, desmina, queratina y HMB-455,12. Clínicamente, nuestro caso parece corresponder más a un FAS. Sin embargo, la infiltración del tejido celular subcutáneo con un patrón en «panal de abeja», la presencia de la translocación t(17;22)(q22;q13) por FISH y la ausencia de células gigantes multinucleadas, presentes en el FAS, lo descartan5,12.
Otros tumores como el fibroma digital celular son fácilmente descartables, ya que clínicamente se presentan como una pápula y no como una masa lobulada, son infrecuentes en niños e histológicamente no se han descrito casos de predominio mixoide13. Son positivos para CD34 y para el factor XIIIa, negativo en nuestro caso. Algunos creen que esta entidad pueda corresponder más bien a una variante celular del fibromixoma acral superficial13.
El angiomixoma superficial es otro tumor que frecuentemente recidiva localmente, aunque no metastatiza. Es de lento crecimiento, se localiza en el tejido subcutáneo, con extensión a la dermis, pero a diferencia de nuestro caso tiene un crecimiento lobular, sin infiltración difusa en «panal de abeja»1. Está constituido por células fusiformes y estrelladas en un estroma basofílico abundante y muy vascularizado. Posee neutrófilos alrededor de estructuras vasculares de paredes finas y elementos epiteliales (cordones epiteliales o quistes de queratina) hasta en un 50% de los casos; ambas características no se observaron en nuestro caso1,3. También es positivo para CD34, vimentina y actina, esta última negativa en nuestro caso y es negativo para desmina, S-100 y factor XIIIa5.
El fibroblastoma de células gigantes es un tumor de niños, con predominio en el sexo masculino, que constituye un espectro morfológico con el DFSP, con el que comparte perfil histológico, inmunohistoquímico y genético. En casi un 15% de los casos de DFSP se encuentran áreas tipo fibroblastoma de células gigantes. Afecta a la dermis y el tejido celular subcutáneo, rodeando los anejos4,6. Las lesiones son hipocelulares, con fascículos laxos de células fusiformes onduladas, en un estroma abundante colagenoso o mixoide, altamente vascularizado. Posee una población característica de células uninucleadas pleomórficas o gigantes multinucleadas, ausentes en el caso que se presenta. Es CD34 positivo y posee la translocación t(17;22)(q22;q13). Puede recidivar como un DFSP y viceversa4.
El perineuroma extraneural de células fusiformes puede tener un estroma mixoide prominente, pero es bien circunscrito a diferencia del borde infiltrativo de nuestro caso y está compuesto por células fusiformes, que se disponen en un patrón en «rueda de carro», lamelar o fascicular. El 50% de los casos son positivos con el CD34, pero a la vez son positivos para marcadores perineurales como Glut-1 y EMA, este negativo en nuestro caso1. Por último, el tumor fibroso solitario aunque es también CD34 y CD99 positivo y puede tener estroma mixoide prominente, es bien circunscrito, expresa bcl-2 y presenta un característico patrón vascular hemangiopericitoide, lo que lo diferencia del presente caso1.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


