





La epidermólisis ampollosa distrófica pruriginosa (EADP) es un subtipo infrecuente de EAD. Se origina por mutaciones del gen COL7A1, que codifica para el colágeno VII, produciendo una disfunción de las fibras de anclaje de la unión dermoepidérmica. Histológicamente se observan ampollas subepidérmicas y clínicamente unas placas liquenificadas muy pruriginosas, habitualmente pretibiales.
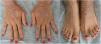
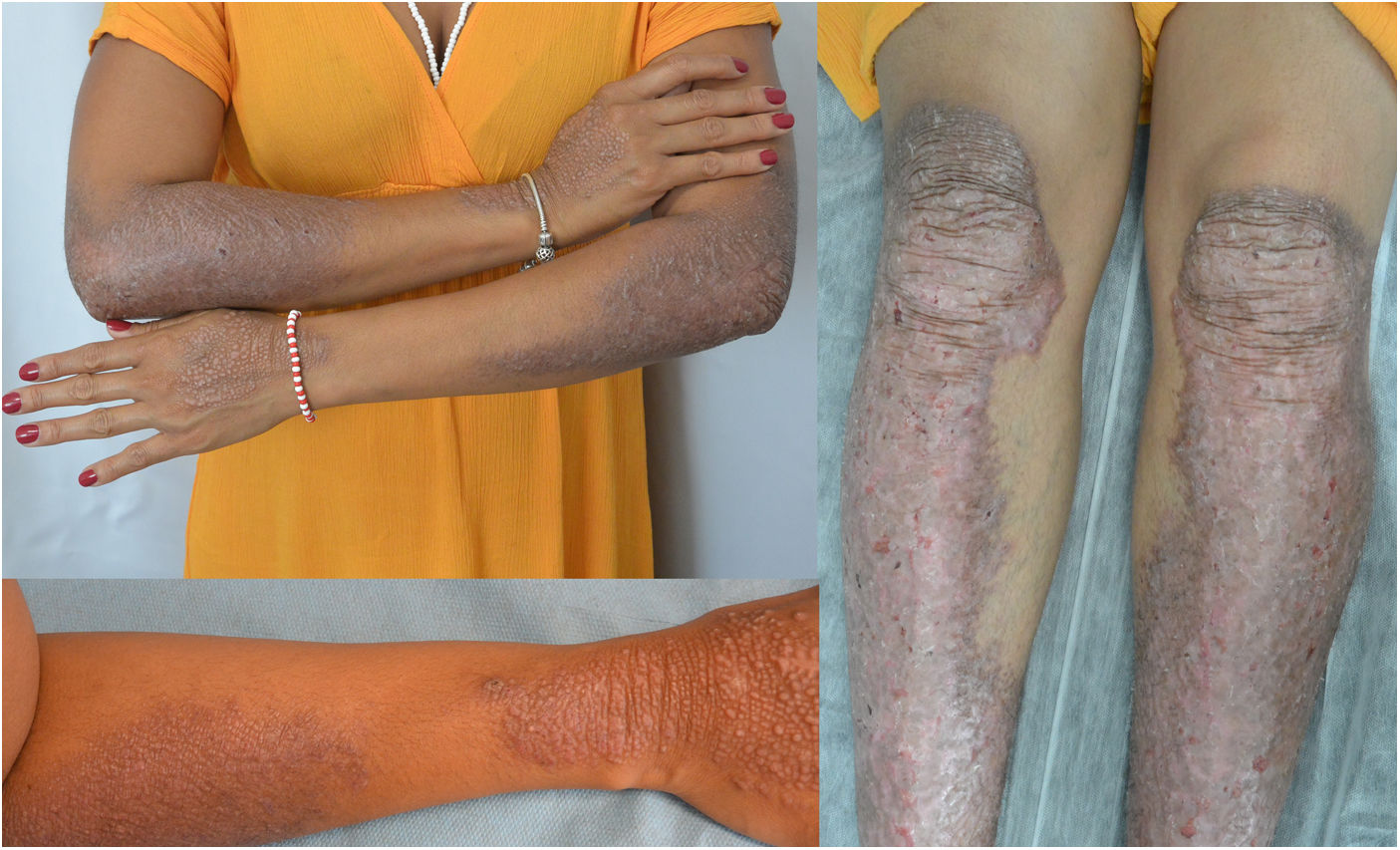
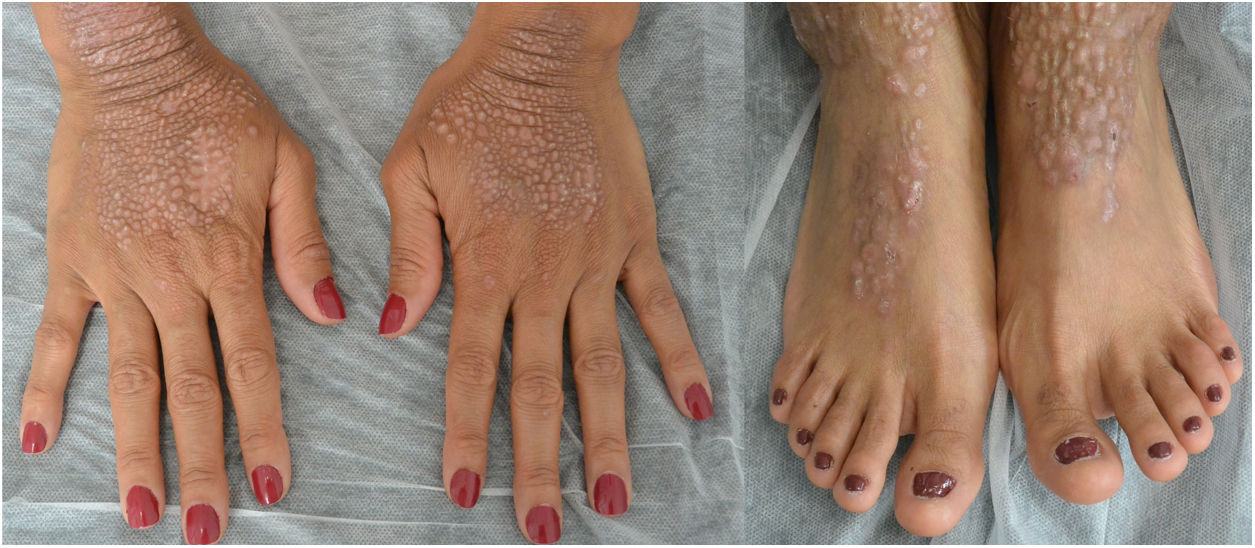
Una mujer de 34 años, sin antecedentes de interés, consultó por unas lesiones intensamente pruriginosas, que presentaba desde los 3 años, localizadas en el dorso de las manos, los antebrazos y en la cara anterior de piernas. Refería la aparición ocasional de vesículas y ampollas, especialmente tras traumatismos. Asimismo, refería un hermanastro por vía materna con unas lesiones similares, pero ni sus padres (no consanguíneos) ni su hija estaban afectados. En la exploración se objetivó la presencia de pápulas y placas liquenificadas marronáceas bien delimitadas con erosiones múltiples en la zona distal de las extremidades, así como distrofia ungueal en los dedos de los pies (figs. 1 y 2).
El estudio histológico de una biopsia mostró la presencia de ampollas subepidérmicas con poca inflamación y una inmunofluorescencia directa negativa para inmunoglobulinas y complemento. La microscopía electrónica evidenció focos de clivaje bajo la lámina densa con una pérdida de las fibrillas de anclaje dérmico. En la analítica solo destacaba una elevación de la IgE (800 UI/ml). Con estos hallazgos la paciente fue diagnosticada de EADP.
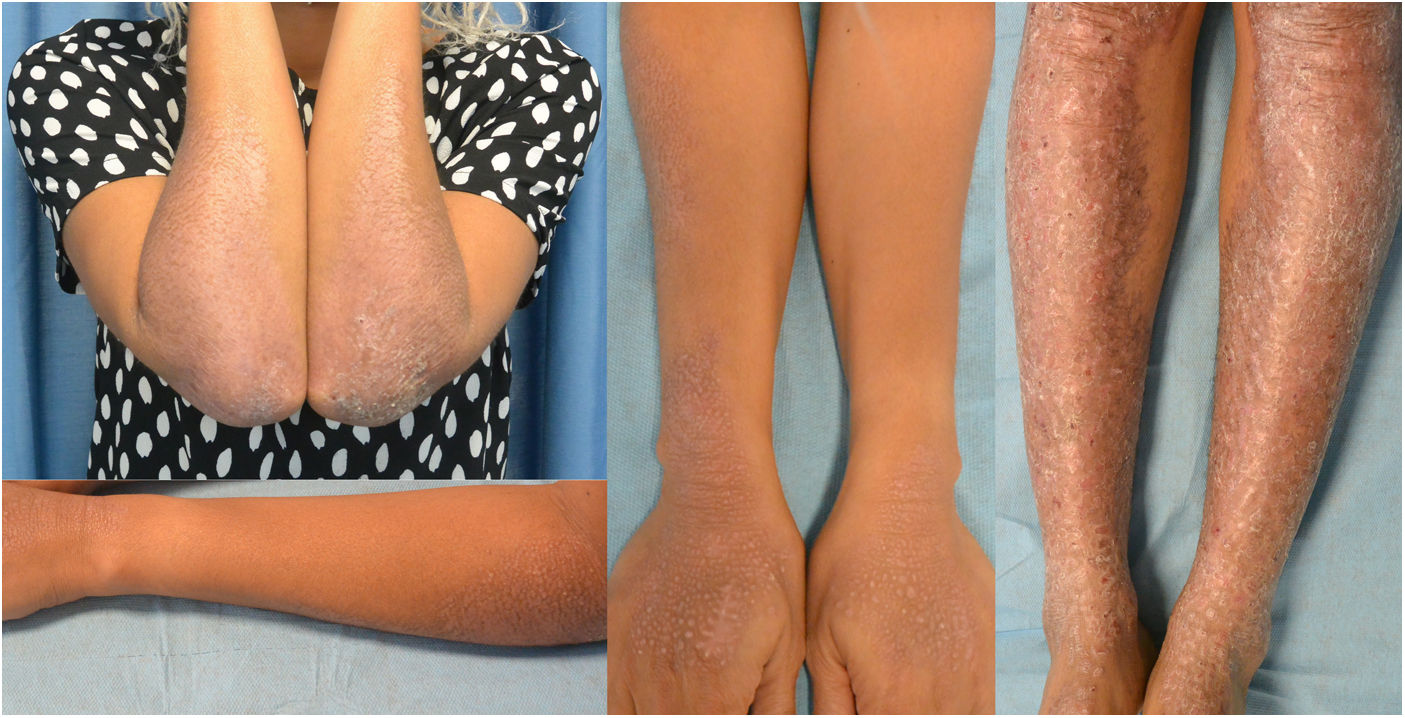
La paciente había sido tratada con múltiples tratamientos tópicos (corticoides, inhibidores de calcineurina, análogos de la vitamina D, fórmulas magistrales de doxepina y amitriptilina) y fármacos sistémicos (talidomida, ciclosporina, gabapentina, mirtazapina y diversos antihistamínicos orales) sin una mejoría del prurito ni de las lesiones. Dada la marcada afectación de la calidad de vida, con un DLQI de 13, se administró dupilumab por uso compasivo a una dosis inicial de 600mg seguida de 300mg cada 2 semanas. A los 14 días la paciente refería un marcado alivio del prurito y, tras 2 meses de tratamiento, se objetivó una mejoría de las lesiones cutáneas. A los 9 meses de tratamiento presentaba una gran mejoría de la calidad de vida, con un DLQI de 5, y de las lesiones cutáneas, sobre todo de los miembros superiores (fig. 3). El único efecto secundario fue una leucopenia leve inicial que se corrigió espontáneamente sin precisar una reducción de dosis.
La EADP fue descrita en 1994 por McGrath1. Es una enfermedad rara de transmisión generalmente autosómica dominante, aunque se han descrito casos esporádicos y de transmisión recesiva. La mutación más frecuente del gen COL7A1 es la sustitución de glicina, sin establecerse hasta la fecha correlación geno-fenotípica2. Dada la gran variabilidad clínica, en ocasiones incluso en la misma familia, se ha hipotetizado que factores como la atopia, el déficit de hierro o una elevación de la IgE puedan actuar como modificadores. Se han descrito 3 casos tras escabiosis3.
El comienzo es variable, habiendo casos tras los 70 años4, aunque habitualmente comienza durante la adolescencia con un prurito incoercible de predominio en las zonas distales de las extremidades que, secundariamente, ocasiona el desarrollo de pápulas, nódulos y placas liquenificadas, que pueden mostrar una distribución lineal, excoriaciones y cicatrices. Puede haber afectación troncular, pero es característico que no se afecten la cara ni las flexuras y que el prurito de mayor intensidad se dé en zona pretibial. Es frecuente la aparición de ampollas tras traumatismos y el hallazgo de distrofia ungueal en pies. Se han descrito 3 casos asintomáticos5.
La histopatología muestra una hiperqueratosis con acantosis y ampollas subepidérmicas respeto a la membrana basal, que quedaría localizada en la porción epidérmica de las mismas, acompañadas de un infiltrado linfocítico intersticial y perivascular. La inmunofluorescencia directa es positiva para el anticuerpo del colágeno VII, con una tinción lineal a lo largo de la membrana basal. La microscopía electrónica muestra, como en otras EAD, alteración de la unión dermoepidérmica y reducción del número de fibras de anclaje. Aunque la prueba confirmatoria sería el estudio genético, es posible su diagnóstico si se dan una clínica característica, la presencia de antecedentes familiares y una histopatología y microscopía electrónica compatibles6.
El diagnóstico puede requerir un alto nivel de sospecha, dado que algunos pacientes comienzan en la edad adulta con escasas lesiones y siendo inusual el hallazgo de ampollas intactas7. El diagnóstico diferencial ha de establecerse principalmente con el prúrigo nodular, el liquen simple crónico y el liquen plano hipertrófico, siendo de ayuda la histopatología.
Se han empleado corticoides tópicos, intralesionales y orales, inhibidores de la calcineurina tópicos, crioterapia, fototerapia UVB, antihistamínicos y retinoides orales, dapsona, gabapentina, talidomida, ciclosporina, mizoribina, tofacitinib, baricitinib, inmunoglobulinas intravenosas, naltrexona, mirtazapina y sertralina, muchos de ellos sin mejoría o con casos exitosos aislados. El dupilumab es un anticuerpo monoclonal humanizado que inhibe la subunidad alfa del receptor de interleucina-4. Está aprobado para la dermatitis atópica, aunque se ha usado con éxito en prúrigo nodular, que comparte características clínicas con la EADP. Hay 6 casos descritos de pacientes tratados con dupilumab, todos ellos con una mejoría del prurito y de las lesiones cutáneas, sin efectos secundarios. Solo un paciente precisó de un incremento de la dosis a 300mg/semana8.
Presentamos un caso de EADP refractario a múltiples tratamientos que presentó mejoría de la calidad de vida y de las lesiones con dupilumab. Recordamos la importancia de sospechar esta entidad en paciente con lesiones pruriginosas de predominio en piernas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los doctores Daniel Cameselle y Rafael Camacho Galán, así como al Servicio de Anatomía Patológica del Hospital Dr. José Molina Orosa, por su inestimable ayuda en el diagnóstico de este caso.


