

Ehlers-Danlos syndrome (EDS) is a heterogeneous group of congenital connective tissue diseases caused by mutations in genes involved in the synthesis or processing of collagen fibers.1 The phenotypic manifestations of EDS vary greatly, and mild cases can go unnoticed until late in life. We present the case of a girl who was diagnosed with classic EDS in our hospital based on clinical and ultrasound findings.
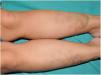
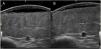
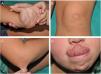
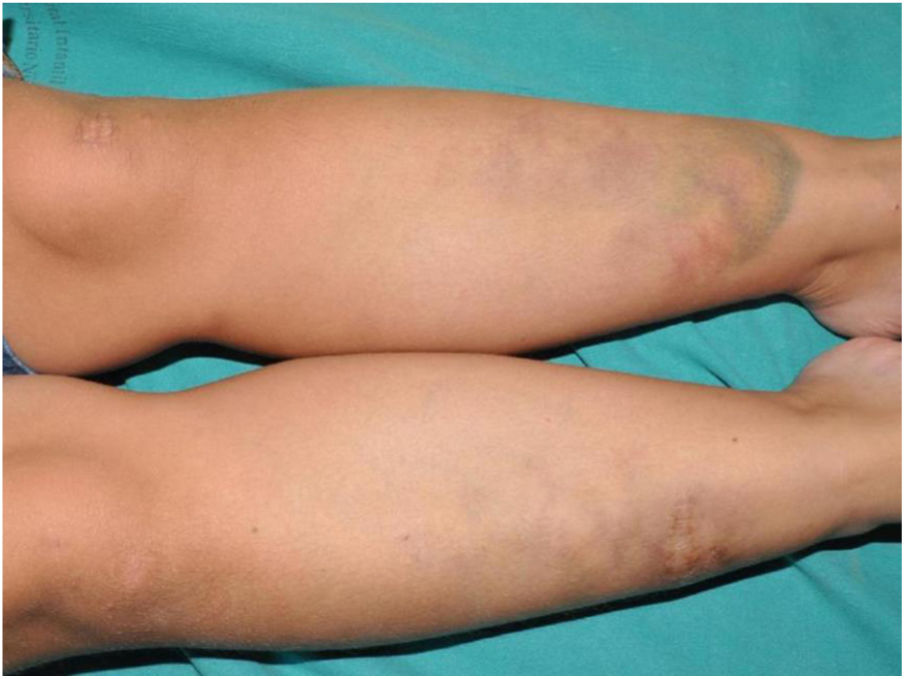
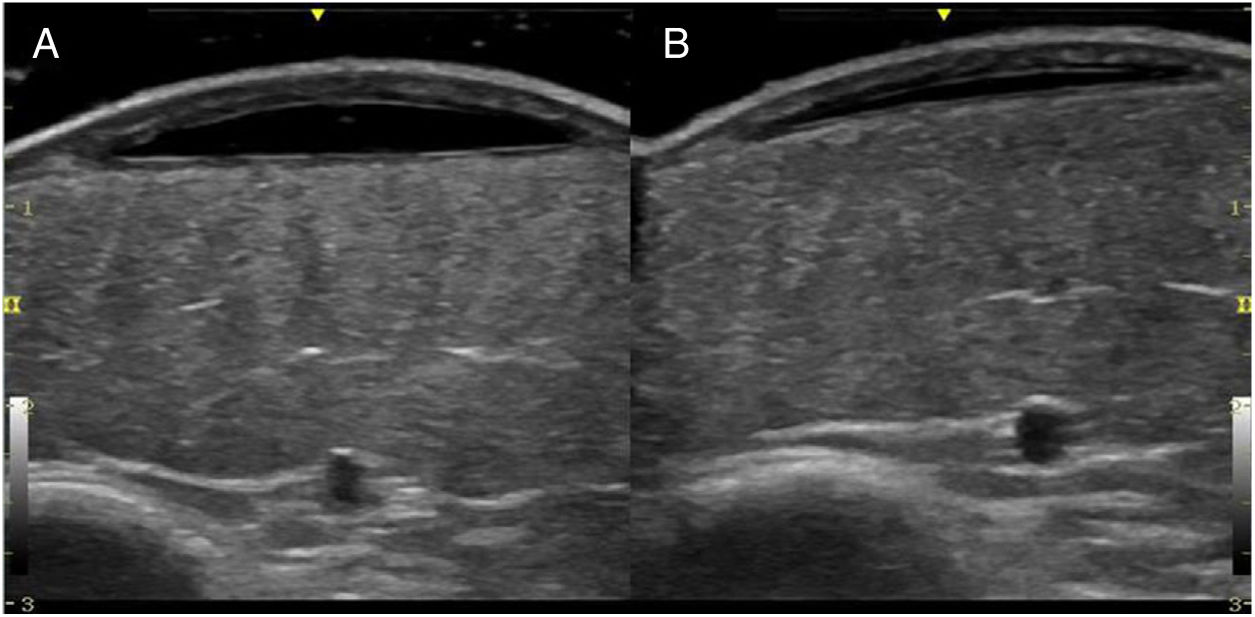
An 8-year-old girl was referred from the emergency department for evaluation of a painful lesion on the left leg that had appeared several weeks earlier after a fall from a ladder. Physical examination revealed a subcutaneous bulge of about 4cm in diameter in the left pretibial region with yellowish-purpuric overlying skin and a strikingly gummy consistency. The patient had reticulated erythematous-violaceous lesions on the right leg and dehiscent and atrophic scars on the right leg and left knee (Fig. 1). High-frequency ultrasound (18 MHz) of the left pretibial lesion revealed an anechogenic collection delimited by a thin pseudocapsule, compatible with an organized hematoma (Fig. 2A). Septal edema compatible with traumatic panniculitis was evident in the surrounding hyperechogenic subcutaneous tissue. Doppler signal was absent. In the directed anamnesis the patient's mother reported that the girl had been born preterm due to premature rupture of membranes and had muscular hypotonia during the neonatal period. The patient was undergoing tests in the endocrinology department of another hospital for short stature and disproportion between the trunk and limbs. The family history provided by the mother included joint hyperlaxity, abnormal scarring, and early osteoarthritis. The patient also presented with skin hyperextensibility, joint hypermobility, and Gorlin sign (ability to reach the nose with the tip of the tongue) (Fig. 3). Based on these data a suspected diagnosis of classic EDS was established. A cardiological examination, including electrocardiogram and echocardiography, revealed no findings of interest, and the results of a laboratory workup, including a complete blood count and coagulation tests, were normal apart from slightly elevated D-dimer levels. A second ultrasound examination performed 1 month later revealed a reduction in the hematoma of approximately 50% (Fig. 2B).
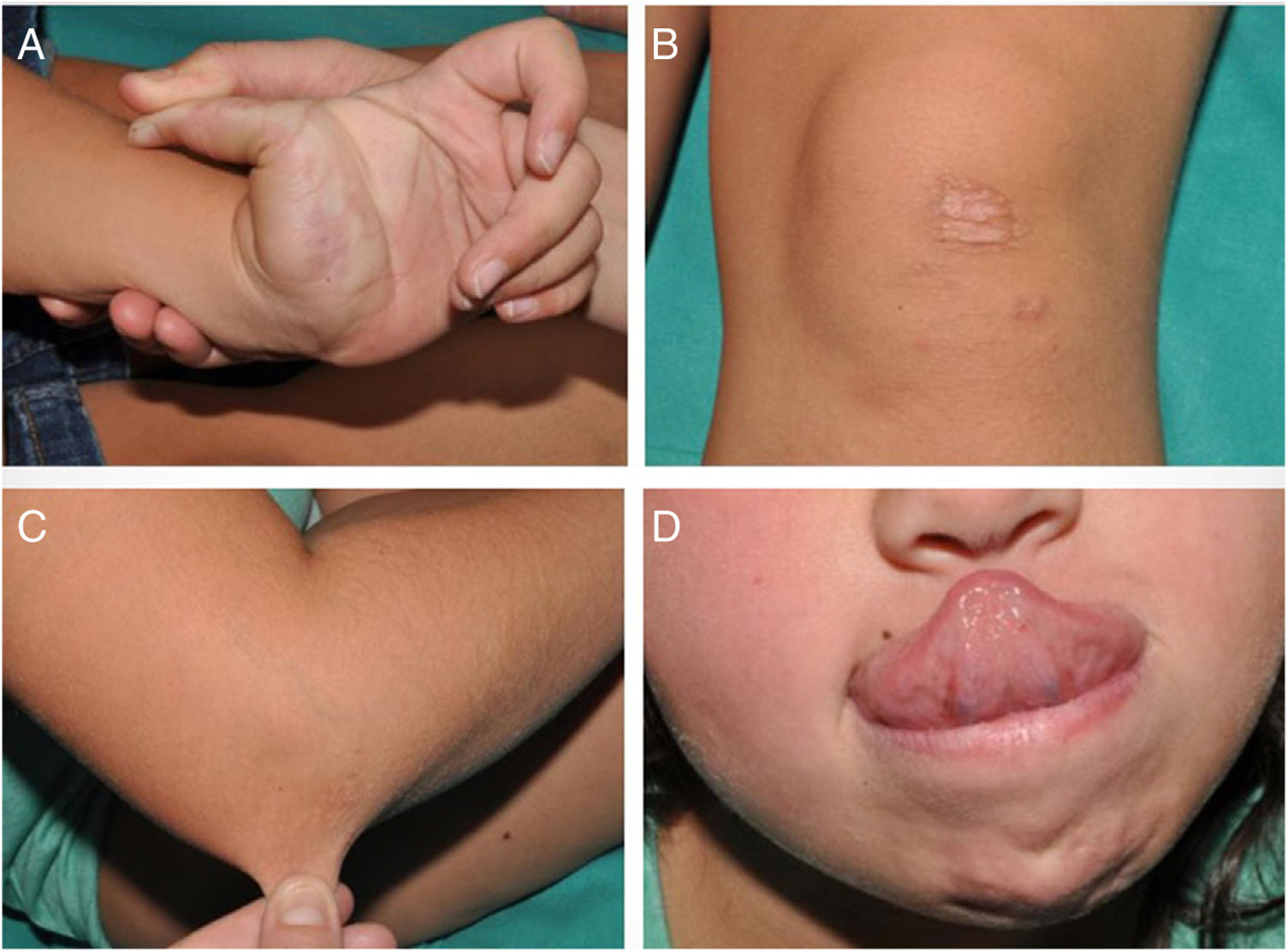
Classic EDS is inherited in an autosomal dominant manner and is caused by mutations in COL5A1 or COL5A2, which encode the alpha-1 and alpha-2 chains, respectively, of collagen type V.1 The disease is characterized by skin hyperextensibility, joint hypermobility and associated complications (luxations, pain, early osteoarthritis), and other clinical characteristics that are reviewed in a recent international consensus paper.1 Dermatologic manifestations of classic EDS include skin hyperextensibility and abnormal scarring, which results the formation of atrophic scars with a cigarette-paper-like appearance. Patients have thin, velvety skin that bruises in response to minimal trauma. Other potential lesions include nodular molluscoid pseudotumors secondary to calcification and fibrosis of hematomas; spheroids (hard spherical nodules on the forearms and pretibial areas); and piezogenic pedal papules.2,3 Our patient fulfilled the diagnostic criteria (Table 1). No genetic study was performed as this was not essential for confirmation of the diagnosis. Classic EDS is usually diagnosed when affected individuals begin to stand and walk, as this period coincides with the appearance of lacerations and bruising that tend to alarm parents. The main clinical differential diagnosis in childhood is bruising caused by child abuse, potentially distinguishing features of which include lesions located in areas not exposed to accidental trauma (e.g., backs of the legs or abdomen), peculiar morphology (e.g., imprints caused by fingers, the dental arch, belts, etc.), and the time course of lesion appearance.4,5 In rarer cases extensive bruising may constitute the initial manifestation of coagulopathies that can be detected by laboratory analyses.
Diagnostic Criteria for Classic Ehlers-Danlos Syndrome
| Major Criteria | Minor Criteria |
|---|---|
| 1. Marked skin hyperextensibility and atrophic scarring2. Generalized joint hypermobility | 1. Easy bruising2. Soft, velvety skin3. Skin fragility4. Molluscoid pseudotumors5. Subcutaneous spheroids6. Hernia (or history thereof)7. Epicanthic folds8. Complications associated with joint hypermobility (e.g., sprains, subluxation/dislocation, pain, flat feet)9. First-degree relative who fulfils the clinical criteria |
For diagnosis it is necessary to fulfil major criterion 1+major criterion 2, or major criterion 1+3 of the minor criteria. Source: Malfait et al., 2017.7
High-resolution ultrasound is a useful tool for diagnostic confirmation of bruising and differentiation from other causes of subcutaneous lesions on the legs, including erythema nodosum, abscesses, and benign (lipoma) or malignant (rhabdomyosarcoma, fibrosarcoma) soft-tissue tumors.6 Hematomas present as anechoic collections that may become hypoechoic or heterogeneous over days or weeks, without peripheral hypervascularization. These lesions can be compressed with the ultrasound probe only in early stages, after which the fluid within is replaced with fibrous tissue. Another feature, which is less well characterized in the literature, is the presence of a thin pseudocapsule that corresponds to the fibrin and peripheral granulation tissue produced as the hematoma organizes. In contrast to hematomas, the aforementioned subcutaneous lesions tend to have more irregular borders and usually differ in terms of the degree of vascularization. Ultrasound also allows measurement of the depth of the hematoma and confirmation of its regression, allowing distinction from hemorrhagic soft-tissue tumors.6
In conclusion, classic EDS should be suspected in children with joint and skin hyperlaxity and easy bruising. High-resolution ultrasound is very useful for the diagnosis and follow-up of hematomas associated with EDS.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Herrero-Moyano M, Noguera-Morel L, Torrelo A, Hernández-Martín A. Síndrome de Ehlers-Danlos clásico: hallazgos clínicos y ecográficos. Actas Dermosifiliogr. 2020;111:83–85.


