












MATERIAL AMILOIDE
El amiloide es un término acuñado por Schleiden en 1838 para describir una sustancia presente en las plantas semejante a la celulosa. Rokitansky, en 1842, realizó la primera descripción morfológica de amiloidosis1 . Los depósitos del amiloide están constituidos básicamente por tres elementos: el amiloide P, las proteínas fibrilares del amiloide y los componentes de la matriz extracelular.
Amiloide P. Es una proteína no fibrilar idéntica a una globulina plasmática circulante normal denominada «amiloide P sérico»2 . Constituye hasta el 14 % del peso del amiloide. Es un componente común a casi todos los tipos de amiloide, independientemente de la proteína precursora. Se cree que actúa como esqueleto básico sobre el cual se depositan las proteínas fibrilares y aunque no se conoce exactamente su función, al ser un inhibidor de elastasas, puede ser que contribuya a proteger los depósitos de la degradación y fagocitosis. Se ha encontrado en suero normal, en suero amiloideo y también en la membrana basal de los glomérulos3 .
Proteínas fibrilares del amiloide. Son diferentes dependiendo del tipo de proteína precursora. Son normales en la población (cadenas ligeras de inmunoglobulinas, prealbúmina, hormonas polipeptídicas, etc.), y no se conoce cuál es el mecanismo por el que se vuelven amiloidogénicas. Estas proteínas presentan unas características comunes fisicoquímicas que les hacen tener las propiedades amiloideas. Aunque las moléculas precursoras intactas completas pueden ocasionalmente formar las fibrillas de amiloide in vivo , generalmente están constituidas por fragmentos que han sufrido una escisión parcial proteolítica. Hasta la fecha, se han reconocido al menos 21 proteínas diferentes como agentes causales de enfermedades amiloideas4 .
Componentes de la matriz extracelular. Formados principalmente por glucosaminoglucanos como heparán sulfato y dermatán sulfato, unidos de forma no covalente a las fibrillas. Su función no está clara, pero parece que tienen una acción fibrilogénica sobre determinadas proteínas precursoras de las fibrillas de amiloide.
CONCEPTO Y CLASIFICACIÓN
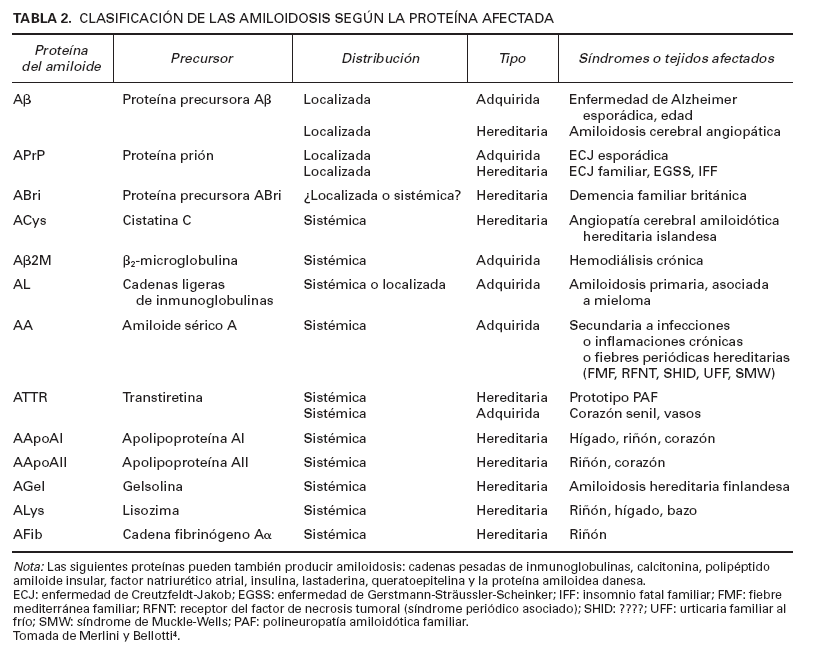
Las amiloidosis son un grupo heterogéneo de enfermedades caracterizadas por el depósito extracelular en los órganos y tejidos de una serie de proteínas fibrilares, no relacionadas bioquímicamente entre sí, con unas características comunes que incluyen la birrefringencia verde manzana con la luz polarizada, previa tinción con rojo Congo, y la configuración en hoja plegada -laminar por difracción de rayos X5-9 . Existen muchas clasificaciones de las amiloidosis. Clásicamente se han clasificado según el cuadro clínico10 como se describe en la tabla 1. Si el depósito de amiloide afecta a muchos órganos se denomina amiloidosis sistémica, y si se localiza en un único tejido se denomina amiloidosis localizada o limitada a un órgano. Recientemente la tendencia es a clasificarlas según la naturaleza de las proteínas precursoras de las proteínas fibrilares del amiloide4,11 (tabla 2), más que por sus características clinicopatológicas.

TABLA 1. CLASIFICACIÓN DE LA AMILOIDOSIS SEGÚN LA ENFERMEDAD
El patrón de depósito de amiloide, y por tanto la sintomatología, se correlaciona con el tipo de proteína precursora que, a su vez, depende de la enfermedad subyacente.
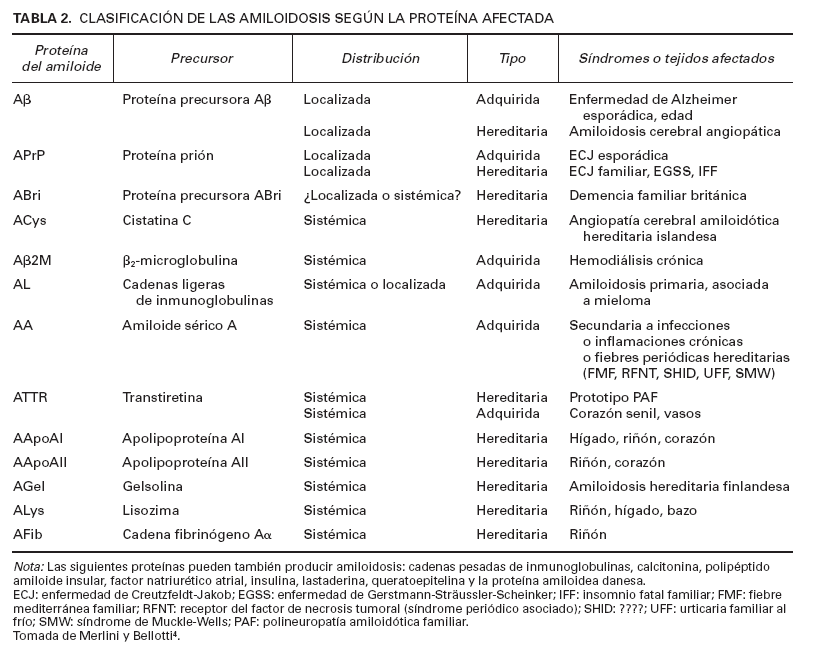
TABLA 2. CLASIFICACIÓN DE LAS AMILOIDOSIS SEGÚN LA PROTEÍNA AFECTADA
AMILOIDOSIS SISTÉMICA ASOCIADA A DISCRASIA DE CÉLULAS PLASMÁTICAS
La amiloidosis sistémica asociada a discrasia de células plasmáticas incluye dos tipos: primaria y asociada a mieloma múltiple. Existe una gran superposición entre ambas formas en la literatura médica. Realmente son parte de un mismo espectro. Es útil diferenciarlas, por el valor pronóstico que de ello se deriva.
Primaria. Se encuentra relacionada con una discrasia de células plasmáticas oculta. Con frecuencia existe una población aumentada de células plasmáticas en la médula ósea, pero no cumplen criterios de mieloma múltiple. No está claro si estos pacientes lo desarrollarán o no.
Asociada a mieloma múltiple. Se da en aproximadamente el 5 al 15 % de los pacientes con mieloma múltiple12 .
PatogeniaParece claro que las proteínas precursoras del amiloide AL son cadenas ligeras de inmunoglobulinas monoclonales enteras y/o sus fragmentos (principalmente la región variable aminoterminal) procedentes de una proliferación clonal de células plasmáticas. En la amiloidosis AL, la sustitución de los aminoácidos en posiciones específicas de la región variable desestabiliza potencialmente las cadenas ligeras y aumenta el riesgo de hacerse amiloidóticas. La mayor parte de las veces, este componente es detectado en sangre u orina, siendo la inmunofijación el patrón de referencia13 . En el 15 % de los pacientes con amiloidosis primaria no se detecta una proteína monoclonal. Esto se explica porque algunas de las proteínas amiloidogénicas están compuestas sólo por fragmentos de 6 a 12 kDa o más pequeñas, a veces sin ninguna secuencia de la región constante, por lo que no pueden ser detectadas por los antisueros policlonales anticadena ligera en estudios inmunohistoquímicos o inmunofluorescentes14 . En estos pacientes se podrían aplicar otras técnicas como la nefelometría15 , Southern blot o reacción en cadena de la polimerasa16 .
La proporción de cadenas ligeras que componen el amiloide suele ser en condiciones normales 3/1 / y en el mieloma múltiple la proporción suele ser 2/3.
Manifestaciones clínicasPresenta un leve predominio en varones, afecta fundamentalmente a edades avanzadas y es excepcional que se manifieste antes de los 40 años. Prácticamente puede afectar a todos los órganos del individuo excepto el cerebro. Típicamente afecta a lengua, corazón, tracto gastrointestinal, esqueleto, músculo liso, ligamentos del carpo, nervios y piel17 (tabla 3).

TABLA 3. SIGNOS Y SÍNTOMAS DE LA AMILOIDOSIS SISTÉMICA
Los síntomas iniciales de presentación son inespecíficos, y es muy difícil el diagnóstico en esta fase. En un estudio de 229 pacientes con amiloidosis primaria la debilidad o fatiga y la pérdida de peso fueron los síntomas iniciales más frecuentes19 . Pueden preceder al diagnóstico histológico incluso en varios años. En cuanto a los síntomas sistémicos, el síndrome del túnel carpiano se presenta hasta en el 25 % de los casos y suele ser rebelde al tratamiento.
Las alteraciones renales son frecuentes, hasta en el 50 %, y son una posible causa de muerte. El depósito se produce, sobre todo, en los glomérulos, aunque también aparecen en los túbulos. El hallazgo más frecuente es la proteinuria, que provoca un cuadro de síndrome nefrótico, y secundariamente edema e hipoalbuminemia. La mayor pérdida de proteínas tiene lugar en áreas de la membrana basal donde ha penetrado el amiloide rompiendo el epitelio20 . La insuficiencia renal se da en grados variables, pero son raras la insuficiencia renal progresiva y la hipertensión arterial.
La afectación cardiaca produce la muerte de hasta el 40 % de pacientes con amiloidosis sistémica19 . El depósito se produce sobre todo en el miocardio, aunque también en el endocardio y el pericardio. La insuficiencia cardiaca congestiva habitualmente es progresiva y de rápida instauración. Puede precederse de alteraciones en el electrocardiograma asintomáticas. Un dato peculiar de la amiloidosis cardiaca es el empeoramiento de la insuficiencia cardiaca tras la utilización de bloqueantes del calcio. Otras manifestaciones menos frecuentes son las arritmias, la angina, el infarto o la hipotensión ortostática, que se pueden producir por infiltración cardiaca21-23 . La combinación de hipertrofia de la pared ventricular, acompañada de un patrón electrocardiográfico de bajo voltaje es muy característica de la amiloidosis cardiaca.
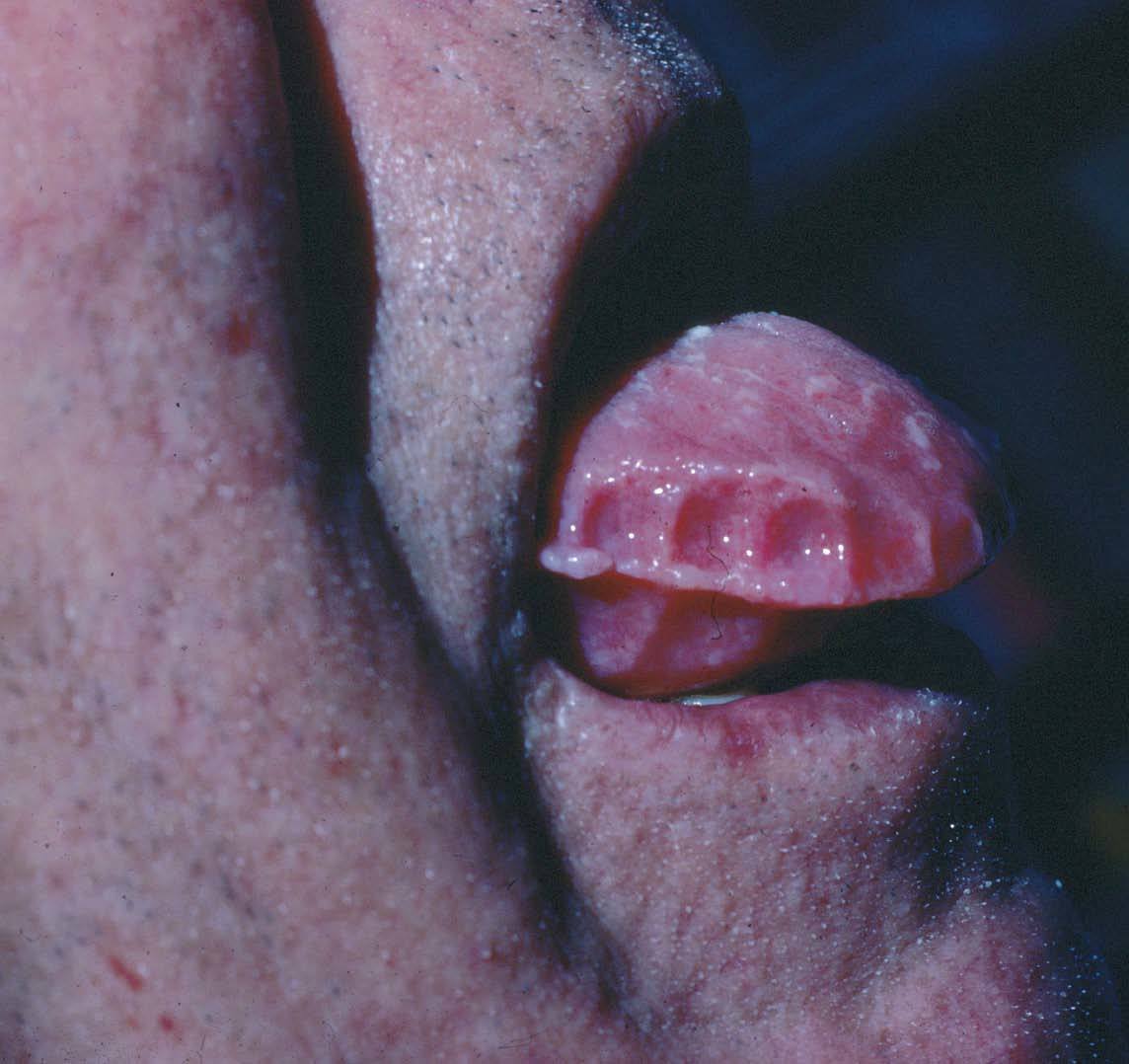
Fig. 1.—Indentaciones en los bordes laterales de la lengua en paciente con amiloidosis sistémica asociada a mieloma múltiple.
También se ve afectado el tracto gastrointestinal pudiéndose producir una hemorragia gastrointestinal, malabsorción, diarrea, ulceraciones, etc. Las alteraciones del sistema nervioso periférico producen principalmente una neuropatía periférica24 , simétrica, con extensión de distal a proximal. Suele ser sensitiva, y con menor frecuencia aparece una neuropatía motora. La afectación se produce sobre todo en los miembros inferiores. Puede acompañarse de una neuropatía autonómica con producción de hipotensión ortostática, diarrea (por alteración de la motilidad gastrointestinal) e impotencia, entre otras. No hay afectación del sistema nervioso central.
La hepatosplenomegalia es secundaria al depósito en bazo e hígado de amiloide, aunque la insuficiencia cardiaca también contribuye a su aparición. La hepatomegalia se observa en el 50 % y la esplenomegalia en menos del 10 %. Es raro que se produzcan alteraciones bioquímicas, aunque con frecuencia la alteración de la función esplénica (hipoesplenismo) se manifiesta por la presencia de cuerpos de Howell-Jolly en el frotis de sangre periférica del 24 % de los enfermos. La linfadenopatía aparece en el 10 % de los pacientes.
La afectación del aparato respiratorio se produce por depósitos en tráquea y bronquios que pueden llegar a provocar signos y síntomas de obstrucción. Estas alteraciones, en ocasiones, son visibles en las pruebas de imagen radiológicas. Por lo general son asintomáticas. La amiloidosis pleural y la afectación pulmonar son raras. Si aparece disnea o derrame pleural suele ser por insuficiencia cardiaca.
Las alteraciones analíticas son diversas y coinciden con los hallazgos descritos en la discrasia de células plasmáticas con paraproteinemia y elevación de la 2-microglobulina en suero.
En los enfermos con amiloidosis sistémica con discrasia de células plasmáticas se han hallado numerosas alteraciones cromosómicas en las células plasmáticas de la médula ósea, incluyendo trisomía de varios cromosomas (7, 9, 11, 18 y X) y monosomía del cromosoma 18, al igual que sucede en el mieloma múltiple y en la gammapatía monoclonal de significado incierto25 . Pueden producirse alteraciones de la coagulación, por déficit aislado del factor X, coagulación intravascular diseminada o fibrinolisis.
Otras manifestaciones de afectación sistémica son claudicación intermitente de miembros inferiores o mandíbula, alteraciones de las glándulas suprarrenales con hipoadrenalismo (hipotensión, hiponatremia), hipotiroidismo (10-20 %), alteraciones de las cuerdas vocales (ronquera y voz apagada), etc.
Las alteraciones cutaneomucosas y de partes blandas se producen en aproximadamente del 21 al 40 % de los casos y con frecuencia son el primer signo de la enfermedad, y son útiles para el diagnóstico precoz. La púrpura, petequias y equimosis son las más habituales; aparecen sobre la piel normal o clínicamente afectada con un tamaño variable. Se producen de forma espontánea o favorecidas por mínimos traumatismos o maniobras de Valsalva. Las localizaciones más frecuentes son los párpados (ojos de mapache), región periorbitaria, axilas, ombligo, área genital, ingles, región submamaria y cuello. Se cree que son consecuencia del depósito de amiloide en la pared de los vasos26 , lo que produciría fragilidad, y de una coagulopatía27 .
La macroglosia se produce en el 19 % de los pacientes con amiloidosis sistémica primaria y en el 32 % de la asociada a mieloma múltiple19 . La lengua está aumentada de tamaño por el depósito del amiloide, generalmente de forma difusa, apareciendo una superficie lisa o abollonada con pápulas, placas o nódulos. Se suelen producir indentaciones en los bordes laterales28 (fig. 1). Suelen formarse lesiones hemorrágicas y, en ocasiones, ampollosas. En algunos casos se describe disfagia y disfonía y rara vez engrosamiento de encías y macroqueilia.
Las pápulas, placas y nódulos con aspecto céreo son las lesiones más características. Se presentan aisladas o con tendencia a confluir formando grandes masas. Presentan un color similar a la piel normal o algo amarillentas. El tamaño es muy variable, desde una cabeza de alfiler a varios centímetros y habitualmente son asintomáticas. La superficie es lisa, son aplanadas y con frecuencia presentan un componente hemorrágico en superficie (fig. 2). Se localizan en las flexuras: párpados, retroauricular, cuello (donde a veces adoptan una morfología tipo pelagroide)29 , axilas, región inguinal y anogenital (donde pueden semejar condi-lomas)30-32 (fig. 3), área central de la cara (donde a veces confluyen y pueden dar lugar a una facies leonina) y labios. También se ha descrito afectación de los conductos auditivos externos33 , vulva (simulando un carcinoma vulvar) 34 , pubis 35 o infiltración cérea de palmas y cara palmar de dedos 36,37 . Otras lesiones cutáneas menos frecuentes se describen en la tabla 4.
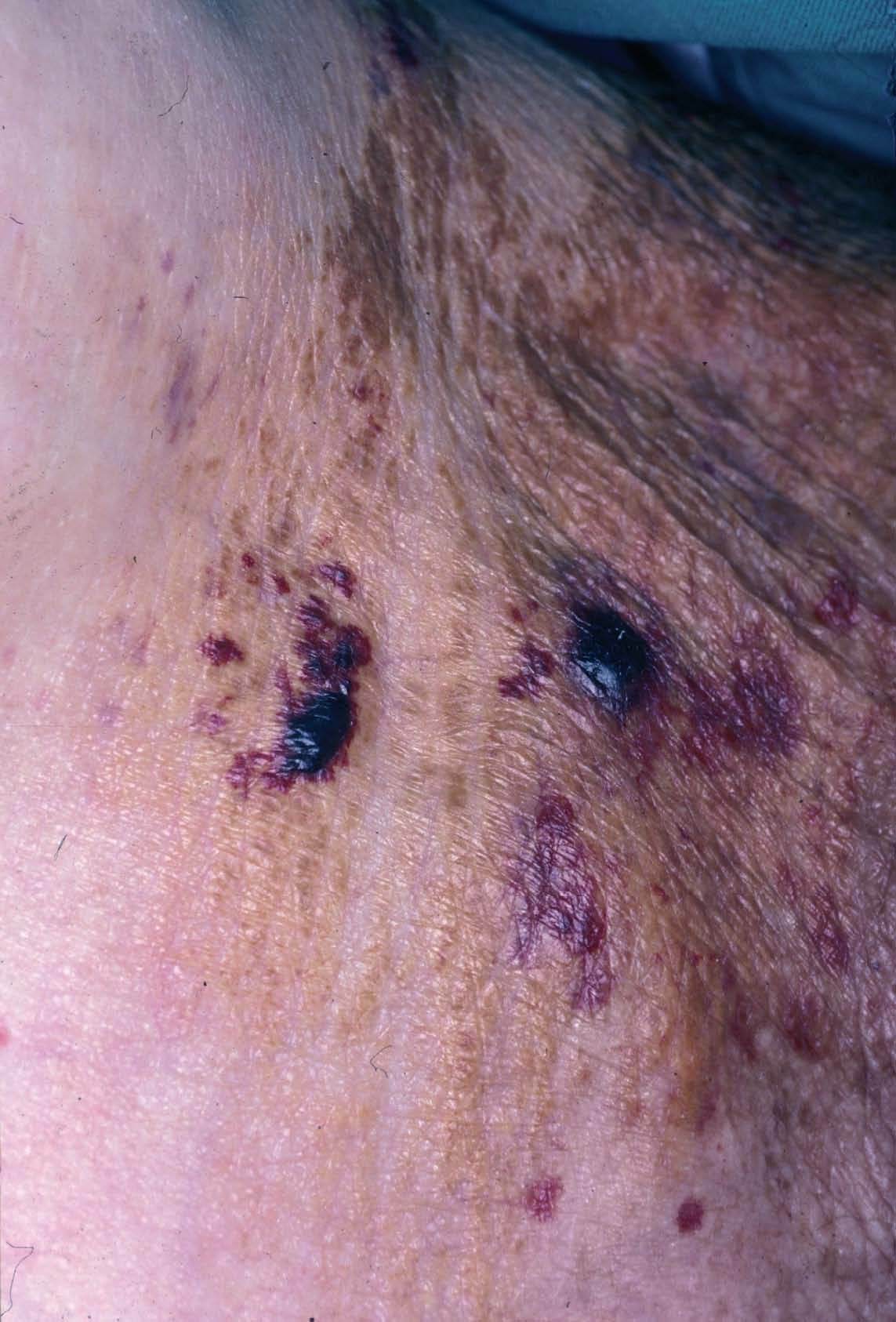
Fig. 2.—Lesiones maculopapulosas de aspecto céreo con hemorragia en superficie.

Fig. 3.—Lesión amiloidea similar a un condiloma.
Diagnóstico
El diagnóstico se basa en la sospecha clínica y la confirmación histológica al observar el amiloide en el órgano afectado. El amiloide, con la tinción de hema-toxilina-eosina, aparece como una sustancia amorfa, eosinofílica, rosada, de aspecto hialino y homogénea (fig. 4). El depósito se realiza fundamentalmente en la pared de los vasos sanguíneos en la dermis y tejido subcutáneo, y a veces también alrededor de glándulas ecrinas y adipocitos. La tinción con rojo Congo (fig. 5) es muy específica y produce una coloración rojo ladrillo, que adquiere un color verde manzana al observarse con luz polarizada. Otras tinciones son tioflavina, metilvioleta, cristal violeta, ácido peryódico de Schiff diastasa, rojo sirio, rojo pagoda, dylon, etc. Además de las tinciones anteriormente expuestas se pueden utilizar técnicas de inmunofluorescencia indirecta e inmunohistoquímica. Otras técnicas incluyen análisis de inmunoabsorción ligado a enzimas 72,73 o Western blot 74 que, utilizando grasa de la pared abdominal o biopsias de tejidos, determinan el tipo de amiloide. Más recientemente, se emplean la espectrometría de masa o el análisis de la secuencia de aminoácidos para la caracterización del depósito proteico según el tipo de amiloide 75,76 .

TABLA 4. MANIFESTACIONES CUTÁNEAS DESCRITAS MENOS FRECUENTEMENTE EN LA AMILOIDOSIS SISTÉMICA ASOCIADA A DISCRASIA DE CÉLULAS PLASMÁTICAS
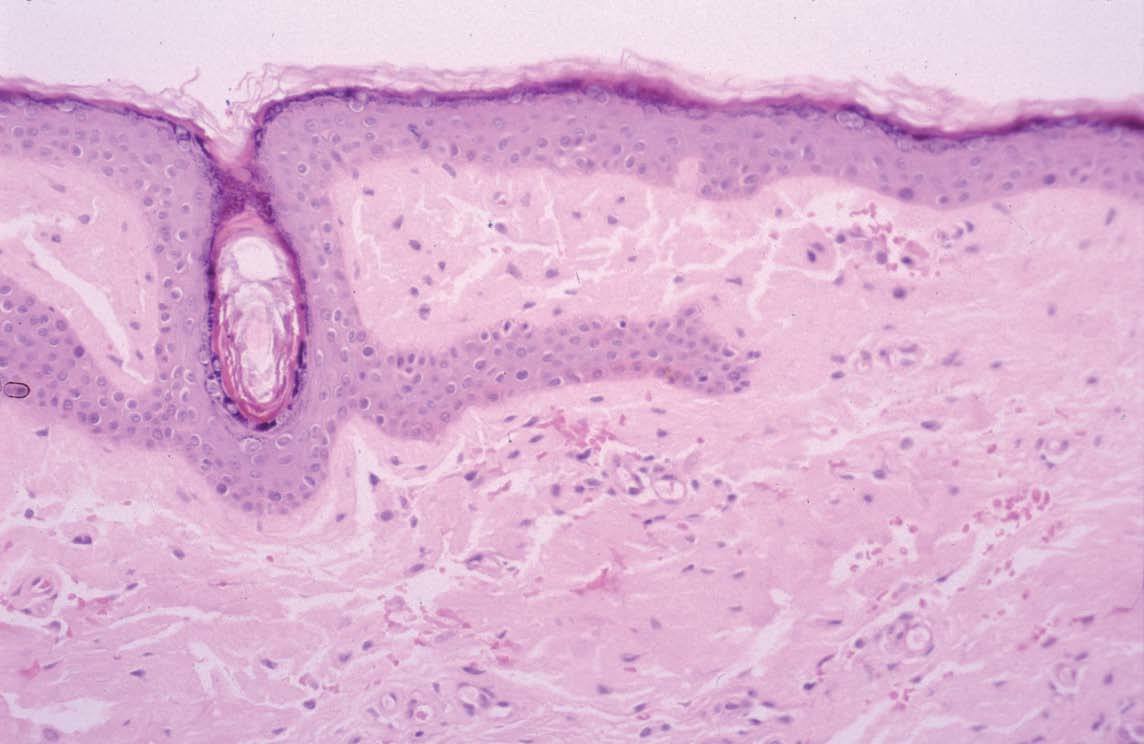
Fig. 4.—Aspecto del amiloide. (Hematoxilina-eosina, ×40.)
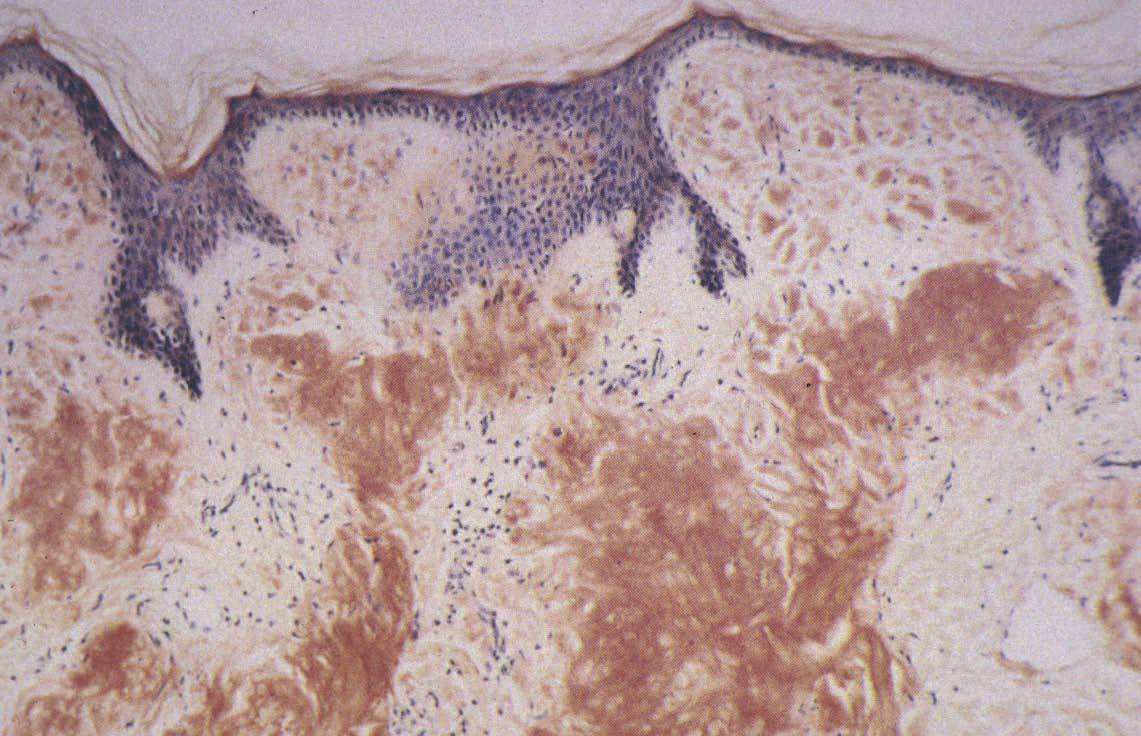
Fig. 5.—Tinción rojo Congo (×50).
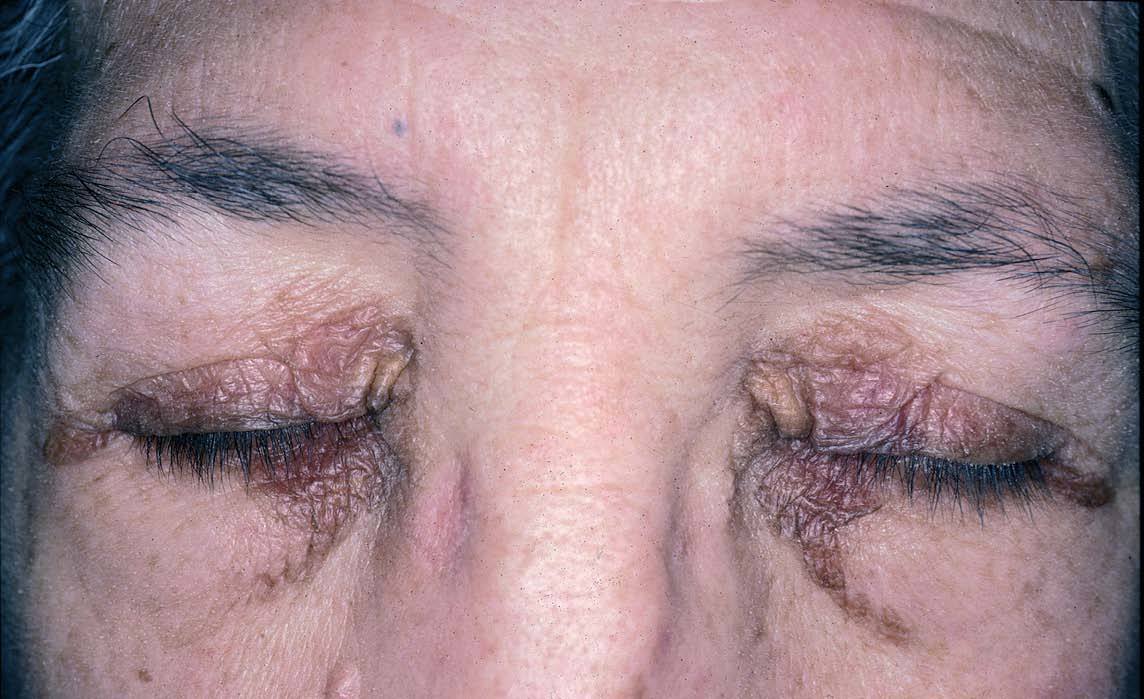
Fig. 6.—Xantelasmas en amiloidosis.
La utilidad de la punción-aspiración con aguja fina (PAAF) de grasa abdominal en el diagnóstico de la amiloidosis sistémica fue descrita originalmente en 1973 por Westermark y Stenkvist77 y posteriormente otros investigadores78-80 la han establecido como una técnica útil para el diagnóstico. La PAAF de grasa abdominal de piel clínicamente normal con aguja fina es un método muy sensible81,82 , sencillo y mínimamente invasivo. Presenta una sensibilidad: del 72 al 95 % en las amiloidosis sistémicas asociadas a discrasias de células plasmáticas83 . Su práctica es útil cuando existe un alto índice de sospecha clínica y no se puede obtener una biopsia de los órganos afectados. La biopsia de piel aparentemente sana del antebrazo, incluyendo tejido subcutáneo, detecta la presencia de amiloide en aproximadamente del 40 al 55 % de las amiloidosis AL84 . Huang et al85 consideran que la biopsia cutánea es más sensible que la PAAF de grasa abdominal, pues en algunos casos el amiloide se localiza en la dermis y no en la grasa subcutánea.
Las técnicas de gammagrafía utilizando el componente amiloide P sérico humano marcado con yo-do-123, yodo-131 o tecnecio-99m, permiten localizar los depósitos de amiloide en el organismo. Se pueden utilizar en los diferentes tipos de amiloidosis. Existe escasa correlación entre la cantidad de amiloide depositado y el grado de alteración funcional en el órgano correspondiente. Sirven para valorar la respuesta al tratamiento86 .
EvoluciónLas amiloidosis sistémicas con discrasia de células plasmáticas, en general, son las que presentan el peor pronóstico. Este depende fundamentalmente del grado de extensión y de la afectación producida por el amiloide. La presencia de insuficiencia cardiaca está asociada con el peor pronóstico y la afectación neuropática al mejor. Las causas más frecuentes de muerte son la cardiaca y la renal. La supervivencia de 810 pacientes con amiloidosis primaria fue del 51 % en un año, 16 % a los 5 años y 4,7 % a los 10 años87 . Recientemente, con métodos terapéuticos actuales, se consiguen supervivencias mayores. Ocasionalmente se han descrito supervivencias de más de 10 años88 .
TratamientoSe ha utilizado la quimioterapia para reducir las células productoras de cadenas ligeras productoras de amiloide16,89,90 . Entre los citostáticos empleados están melfalán, adriamicina, azatioprina, vincristina y ciclofosfamida, que se usan en monoterapia o en diferentes combinaciones. En la mayoría de los tratamientos se añaden esteroides tópicos. La yododesoxirrubicina moviliza los depósitos de AL, aunque no parece aumentar la supervivencia91 . En pacientes menores de 70 años con mieloma múltiple y amiloidosis asociada se puede realizar quimioterapia intensiva y posterior trasplante autólogo de células madre92-95 .
Otras terapéuticas en experimentación son las inmunotoxinas dirigidas contra los precursores de las células plasmáticas amiloidogénicas96,97 y los anticuerpos contra la región variable de la cadena ligera91,98 . El tratamiento sintomático es fundamental, pudiéndose utilizar hemodiálisis y trasplante cardiaco99 o renal.
AMILOIDOSIS SISTÉMICA SECUNDARIA O REACTIVA
La amiloidosis sistémica secundaria o reactiva está asociada a muchas enfermedades, generalmente sistémicas inflamatorias crónicas o agudas recurrentes. Entre ellas, existen numerosas dermatosis descritas en la tabla 5, destacando la psoriasis100-104 . En esta última, son más frecuentes en los casos de larga evolución, la psoriasis pustulosa y sobre todo en la artropatía psoriásica. Se han relacionado los retinoides con la amiloidosis en psoriasis como agentes capaces de movilizar el amiloide. La amiloidosis secundaria se suele diagnosticar como consecuencia del daño tisular en otros tejidos, ya que la afectación cutánea evidente es rara.
PatogeniaLa proteína precursora de la proteína fibrilar del amiloide es una proteína sérica denominada amiloide A sérico (AAS). Se sintetiza principalmente en los hepatocitos y se comporta como un reactante de fase aguda en la población sana, que se produce en respuesta a la inflamación. Se conocen varias proteínas AAS. En humanos, los depósitos de amiloide AA están constituidos por fragmentos de al menos cinco tipos moleculares diferentes108 . El mecanismo patogénico de formación del amiloide AA a partir de AAS no está claro.
Manifestaciones clínicasLa sintomatología es consecuencia del depósito de amiloide en los diferentes tejidos, sobre todo los riñones, hígado, bazo y suprarrenales. Es similar a la amiloidosis AL, pero con mayor frecuencia de afectación renal (tabla 3). La afectación cardiaca raramente es sintomática, pero a menudo se encuentran depósitos en autopsias. Las glándulas suprarrenales se ven afectados en un tercio de los casos, aunque por lo general no se produce alteración de la función.
Las lesiones cutaneomucosas son excepcionales. Brownstein y Helwig58 revisaron 100 casos de amiloidosis AA y ninguno de los pacientes presentaba lesiones cutáneas. Se han descrito petequias y otras lesiones purpúricas en tronco y extremidades84 . Habitualmente no presentan macroglosia y las lesiones ampollosas son excepcionales109 (fig. 7). También se ha descrito aumento de partes blandas en piernas (amiloidomas)110 y alopecia84 .
DiagnósticoSe pueden utilizar las mismas técnicas que en el amiloide AL para demostrar el material amiloide en los órganos con sospecha clínica. La PAAF con posterior tinción con rojo Congo es menos sensible, ya que es positiva en aproximadamente el 66 % de los casos81 , aunque es el método más sensible para el diagnóstico. La biopsia cutánea de piel aparentemente normal y tinción con rojo Congo es positiva en aproximadamente el 50 %84 . El amiloide se localiza predominantemente en dermis profunda, alrededor de vasos, anejos y adipocitos. Es raro ver amiloide en la dermis superficial y media37,111 . El aspecto del amiloide es similar al del amiloide AL, aunque la cantidad suele ser menor (fig. 8).
PronósticoEsta forma de amiloidosis tiene mal pronóstico, si bien depende principalmente de la enfermedad subyacente. Con frecuencia tiene una evolución fatal (media de vida desde el diagnóstico de 4-5 años). La causa más frecuente de muerte es la insuficiencia renal. En un estudio de 80 pacientes seguidos durante largos períodos se sugería que aquellos con un nivel menor de 10 mg/l de proteína AAS tenían una supervivencia mayor que los individuos con niveles superiores112 .

TABLA 5. DERMATOSIS ASOCIADAS A AMILOIDOSIS SECUNDARIA
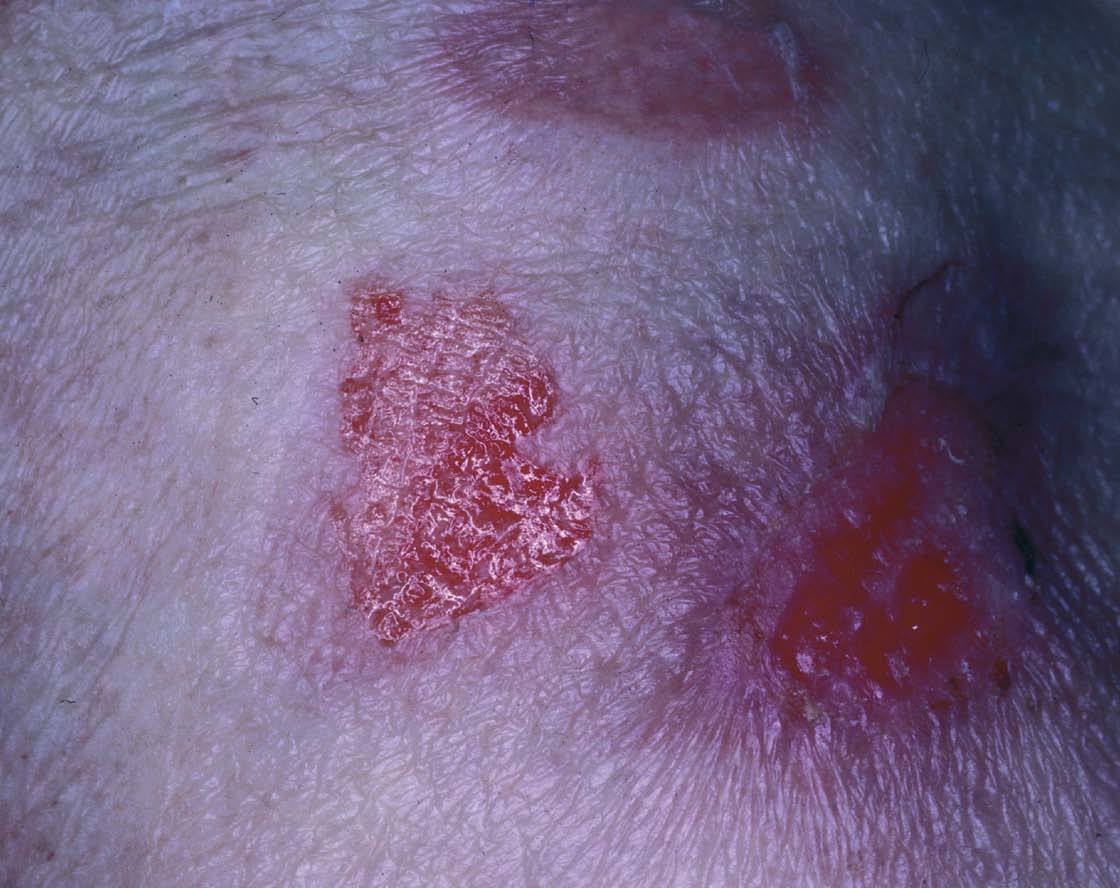
Fig. 7.—Lesión ampollosa en amiloidosis secundaria.
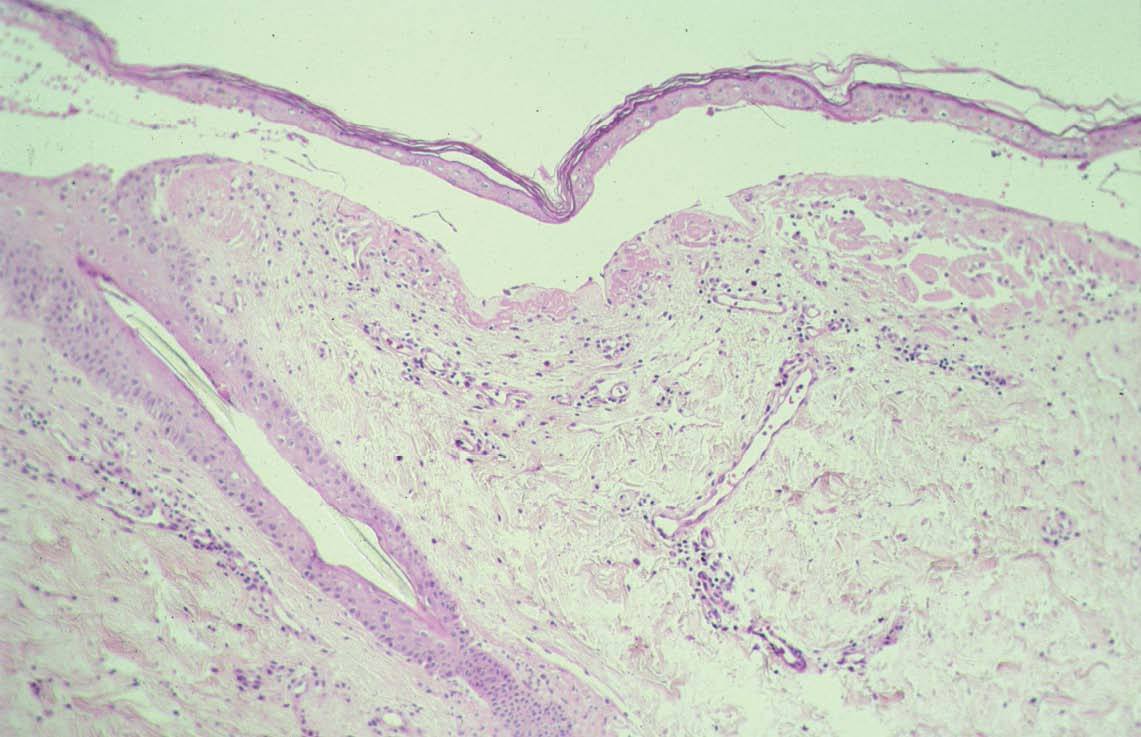
Fig. 8.—Amiloidosis secundaria con ampolla. (Hematoxilina-eosi-na, ×50.)
TratamientoEl tratamiento principal es el de la enfermedad subyacente. Existen numerosos trabajos en la literatura médica en los que desaparece la amiloidosis sistémica secundaria tras el tratamiento de la enfermedad que lo produce89 . El tratamiento de la amiloidosis secundaria a enfermedades reumatológicas con agentes alquilantes quimioterápicos mejora la afectación renal y mejora la supervivencia113 . No existe un tratamiento realmente eficaz. Se ha utilizado el dimetilsulfóxido, que es útil en la afectación renal, se cree que por sus propiedades antiinflamatorias5 . La colchicina mejora la función renal en pacientes con artropatía psoriásica. Al igual que en la amiloidosis sistémica asociada a discrasia de células plasmáticas, es fundamental el tratamiento sintomático y de soporte.
AMILOIDOSIS SISTÉMICA ASOCIADA A HEMODIÁLISIS
Suele aparecer asociada a hemodiálisis crónica (generalmente más de 8 años). Dado que afecta sobre todo a articulaciones de forma aparentemente exclusiva se la suele incluir en las amiloidosis localizadas. Sin embargo, se ha descrito también la afectación de otros órganos114,115 como corazón, mucosa intestinal, lengua, piel, etc.
PatogeniaLa proteína precursora de la proteína fibrilar del amiloide es la 2-microglobulina116 , que se corresponde con el fragmento constante de la cadena de la molécula del complejo mayor de histocompatibilidad de clase I. Está siendo constantemente eliminada a partir de las membranas celulares al torrente sanguíneo. Se filtra en el glomérulo renal y se reabsorbe en el túbulo proximal donde es catabolizada. Estos pacientes presentan niveles altos de 2-microglobulina que no es eliminada por las membranas de celulosa o cuprofano de la diálisis. No se sabe por qué las membranas de celulosa pueden favorecer el depósito, pero parece que resultan menos porosas que las membranas sintéticas actuales. Sin embargo, todavía existen muchos aspectos que no están claros, ya que muchos pacientes en diálisis y con concentraciones elevadas nunca presentan amiloidosis, por lo que deben participar otros factores.
ClínicaEl amiloide se puede depositar en varios tejidos, como el perineural, periarticular, en las articulaciones, piel, hígado, bazo, mucosa intestinal, corazón, riñón, próstata, vasos sanguíneos, etc. Su expresión clínica suele ser bastante limitada, afectando generalmente las estructuras osteoarticulares117 . Se afectan, sobre todo, la membrana sinovial, cápsula articular y discos intervertebrales. Los síntomas más frecuentes son el síndrome del túnel carpiano y la artropatía amiloidea.
Las manifestaciones cutáneas son muy raras. Se han descrito dedos de las manos amigados36,118 , lesiones papulosas liquenoides en el tronco y brazos119 y masas glúteas dolorosas120 . Las lesiones en la lengua suelen corresponder a nódulos de tamaños variables y consistencia firme, sin macroglosia121 . Se trata de lesiones localizadas o difusas por toda la lengua. Es frecuente la alteración del gusto y la dificultad en la movilidad. Matsuo et al122 describieron 8 casos de amiloidosis en lengua en 472 hemodializados.
TratamientoEl cambio de las membranas de la diálisis por membranas sintéticas muy permeables y/o dializados ultra-puros mejora la sintomatología. El trasplante de riñón detiene la progresión de la enfermedad y mejora las manifestaciones clínicas osteoarticulares, pero probablemente no hace desaparecer los depósitos ya instaurados.
AMILOIDOSIS SISTÉMICAS HEREDOFAMILIARES
Son un grupo de amiloidosis muy variado en los cuales una proteína mutante actúa como precursora para formar las fibrillas de amiloide. Existen muchas formas y son poco frecuentes. Suelen manifestarse en la edad media de la vida. Con frecuencia afectan a grupos étnicos (amiloidosis sistémica heredofamiliar tipo finlandés, tipo portugués, etc.). Generalmente son autosómicas dominantes, limitadas a un órgano y rara vez presentan manifestaciones cutáneas.
PatogeniaLas proteínas precursoras son mutaciones de proteínas normales plasmáticas. La más frecuente es la transtiretina65 , que es la proteína de transporte de la tiroxina y de la proteína de unión a retinol123 . Otras son la apolipoproteína AI, gelsolina, -fibrinógeno, lisozima, cistatina C, etc. Se conocen más de 50 sustituciones diferentes de aminoácidos en la transtiretina que pueden causar amiloidosis familiar, sobre todo de formas predominantemente neuropáticas. En estos casos, se ha visto que el trasplante de hígado (lugar de síntesis de la transtiretina) es capaz de hacer desaparecer la proteína mutante de la sangre y mejora las manifestaciones clínicas. No está claro qué sucede a largo plazo.
Manifestaciones clínicasClásicamente se dividen en formas predominantemente neuropáticas, formas predominantemente nefropáticas y formas predominantemente cardiomiopáticas. También puede haber depósito en otros tejidos. Las manifestaciones clínicas tienden a ser similares para cada mutación de una determinada proteína precursora. La macroglosia no es excepcional.
En este apartado destaca la fiebre mediterránea familiar y el síndrome de Muckle-Wells dado que presentan manifestaciones cutáneas.
Fiebre mediterránea familiar
La fiebre mediterránea familiar, también denominada poliserositis familiar paroxística tiene un patrón de herencia autosómico recesivo. A veces existe historia familiar o antecedentes de consanguinidad en familiares. Afecta sobre todo a judíos sefarditas (50 % de los casos), armenios, árabes y turcos.
Recientemente, se ha localizado y clonado el 124 denominado MEFV , que se localiza en el brazo corto del cromosoma 16 y codifica una proteína de 781 aminoácidos denominada pirina (o marenostrina) expresada en los neutrófilos. Se piensa que la pirina puede inhibir la transcripción nuclear de promotores de la inflamación, aunque la patogenia no está clara. La sintomatología125-127 se inicia sobre todo en la infancia, con brotes autolimitados recurrentes de fiebre intermitente que duran 1 o 2 días, acompañados de leucocitosis, aumento de la velocidad de sedimentación y poliserositis (peritonitis, pleuritis, pericarditis y sinovitis).
Las alteraciones cutáneas más características, casi pa-tognomónicas128-130 , son lesiones tipo erisipela en piernas y pies que producen dolor, calor, edema y eritema intensos en la porción más distal de los miembros inferiores. Son placas de bordes bien definidos con un tamaño de 15 a 50 cm. Pueden ser desencadenadas por esfuerzos físicos y ceden de forma espontánea en 48 a 72 h con reposo. También se han descrito relacionados con la fiebre mediterránea familiar, urticaria, púrpura de Schönlein-Henoch127,131 , pioderma, nódulos subcutáneos, lesiones ampollosas, celulitis neumocócica, edema angioneurótico y púrpura inespecífica.
Puede acompañarse en el 25 % de los casos de amiloidosis renal, que generalmente se manifiesta bastantes años después de la aparición y repetición de los episodios de fiebre. El depósito de amiloide es AA132 y realmente es una complicación de los episodios recurrentes, más que una característica del cuadro. Es la causa más frecuente de muerte en estos pacientes.
El diagnóstico es fundamentalmente clínico, ya que no existe ningún marcador bioquímico o serológi-125,133 . Recientemente, las técnicas de genética molecular son capaces de determinar mutaciones del gen MEFV , que se puede confirmar molecularmente en los casos de sospecha clínica y detectar individuos con riesgo134 .
El tratamiento de elección es la colchicina135,136 que previene el desarrollo de la amiloidosis, el deterioro de la función renal y disminuye la frecuencia de los brotes. El interferón inyectado al inicio de los brotes parece ser que los alivia.
Síndrome de Muckle-Wells
Este síndrome fue descrito en 1962 por Muckle y Wells137 en una familia con nueve miembros afectados. La mayoría tienen herencia autosómica dominante, pero hay casos descritos de penetrancia incompleta y formas esporádicas. Recientemente se ha localizado el locus responsable de la enfermedad en el cromosoma 1q44138 . El síndrome de Muckle-Wells se debe a mutaciones en el gen CIAS1139 , que codifica una proteína llamada criopirina. Esta enfermedad es alélica con el síndrome CINCA140 y el síndrome autoinflamatorio familiar por frío141 .
La sintomatología142-145 se caracteriza por brotes de lesiones habonosas de tipo urticaria, evanescentes y recurrentes desde la infancia que duran unas horas, que son habitualmente asintomáticas. No responden a los antihistamínicos y representan casi siempre el signo más precoz. Estos brotes suelen acompañarse de fiebre, escalofríos, malestar general, dolor en extremidades, artralgias y artritis. Con el tiempo puede aparecer sordera progresiva nerviosa perceptiva y amiloidosis multiorgánica AA, sobre todo renal, que con frecuencia lleva a la insuficiencia renal y a la muerte. No obstante, existen formas incompletas sin afectación renal o sordera.
AMILOIDOSIS NODULAR
Aunque no es una amiloidosis sistémica sino localizada, por su composición comentaremos también sus características. Está compuesta por depósitos de amiloide que contienen cadenas ligeras de inmunoglobulinas, especialmente cadenas . Parece que el amiloide se produce localmente por las células plasmáticas del infiltrado, y no en las de la médula ósea. El mecanismo por el cual las células plasmáticas sintetizan localmente el amiloide es desconocido146 . Los depósitos de amiloide son indistinguibles de los de las amiloidosis sistémicas asociadas a discrasia de células plasmáticas.
Estudios de reordenamiento genético han confirmado la clonalidad de las células plasmáticas productoras de amiloide en las lesiones cutáneas, pero no en las células plasmáticas de la médula ósea, sugiriendo que se trate de un plasmocitoma extramedular147 . Sin embargo, Inazumi et al148 caracterizaron la proteína fibrilar del amiloide y comprobaron que era policlonal sugiriendo que, al menos en este caso, el depósito nodular de amiloide era de origen reactivo y no neoplásico.
Manifestaciones clínicasEs la forma menos frecuente de las amiloidosis cutáneas. Afecta más a las mujeres (2/1) en la sexta y séptima décadas de la vida. Clínicamente es indistinguible de la afectación nodular de las amiloidosis sistémicas. Dado que en ocasiones se asocia a discrasia de células plasmáticas, algunos casos pudieran corresponder a formas localizadas cutáneas sin evidencia de afectación sistémica, que posteriormente pudieran extenderse a otros órganos32 . Se presentan como un nódulo o nódulos subcutáneos149-151 eritematosos amarronados, de aspecto céreo y consistencia firme. El número y el tamaño son variables, generalmente entre 1-3 cm. Con frecuencia presentan telangiectasias y hemorragia (púrpura) en superficie; a veces en el centro se aprecia atrofia. La localización es variable apareciendo en cara, extremidades, tronco y genitales y lengua152,153 . Las lesiones son asintomáticas. Generalmente la evolución es benigna durante años, pero a veces existe progresión de lesiones nodulares localizadas a amiloidosis sistémica (menos del 15 %) o aparece una paraproteinemia58,154 . Por ello, es conveniente realizar estudios de inmunoglobulinas, radiografía y médula ósea para descartar paraproteinemia o enfermedad sistémica.
Correspondencia:
Sara B. Álvarez-Ruiz. Servicio de Dermatología. Hospital Universitario de La Princesa. Diego de León, 62. 28006 Madrid. España. agarcia@aedv.es
Recibido el 28 de julio de 2004. Aceptado el 27 de septiembre de 2004.


