





El vitíligo es un trastorno cutáneo adquirido caracterizado por la despigmentación progresiva de la piel debido a la pérdida selectiva de melanocitos. Su etiopatogenia es compleja y multifactorial, involucrando una interacción entre factores genéticos, inmunológicos y ambientales. Las evidencias recientes se inclinan hacia un modelo integrado en el que la autoinmunidad, mediada principalmente por linfocitos T citotóxicos y citoquinas inflamatorias, desempeña un papel central. Además, el estrés oxidativo contribuye significativamente a la disfunción y apoptosis de los melanocitos. Estudios genéticos han identificado numerosos loci asociados con el vitíligo, destacando la participación de genes relacionados con la inmunidad adaptativa e innata, así como con el metabolismo celular. Asimismo, factores ambientales como el trauma, la exposición a sustancias químicas y el estrés psicosocial pueden actuar como desencadenantes en individuos predispuestos. Esta revisión sintetiza los avances más recientes en la etiopatogenia del vitíligo, ofreciendo una visión completa de los mecanismos implicados, y abriendo nuevas posibilidades para el desarrollo de enfoques terapéuticos basados en este conocimiento.
Vitiligo is an acquired skin disorder characterised by progressive depigmentation of the skin due to selective loss of melanocytes. Its aetiopathogenesis is complex and multifactorial, involving an interaction between genetic, immunological and environmental factors. Recent evidence leans towards an integrated model in which autoimmunity, mediated mainly by cytotoxic T lymphocytes and inflammatory cytokines, plays a central role. In addition, oxidative stress contributes significantly to melanocyte dysfunction and apoptosis. Genetic studies have identified numerous loci associated with vitiligo, most notably the involvement of genes related to adaptive and innate immunity as well as cellular metabolism. Environmental factors such as trauma, chemical exposure and psychosocial stress may also act as triggers in predisposed individuals. This review synthesises the most recent advances in the aetiopathogenesis of vitiligo, providing a comprehensive overview of the mechanisms involved and opening up new possibilities for the development of therapeutic approaches based on this knowledge.

El vitíligo es un trastorno cutáneo despigmentante que se caracteriza por la pérdida selectiva de melanocitos. Su prevalencia oscila entre un 0,5 y un 2%, según una extensa revisión de los datos de prevalencia de más de 50 estudios en todo el mundo, sin predilección por género ni raza1.
Durante años, la etiopatogenia del vitíligo ha sido objeto de estudio y, aunque se han propuesto diversas teorías, aún persisten muchos interrogantes sobre sus causas precisas y los mecanismos subyacentes. En este artículo, exploraremos las investigaciones más recientes que arrojan luz sobre la compleja interacción de factores genéticos, autoinmunes, neurogénicos y ambientales que contribuyen al desarrollo y progresión del vitíligo. A través de una revisión exhaustiva de la literatura actual, se pretende ofrecer una visión actualizada de los avances en la comprensión de esta enigmática enfermedad, así como de las nuevas perspectivas terapéuticas que emergen a partir de estos conocimientos.
Mecanismos inmunitarios implicados en la etiopatogenia del vitíligoInmunidad innataEl sistema inmunitario innato se considera el vínculo crucial que conecta el estrés oxidativo con la respuesta inmunitaria adaptativa en el desarrollo del vitíligo. En la piel lesional y perilesional de los pacientes se encuentran células dendríticas, macrófagos, células NK activadas y células productoras de IFN-γ, siendo los melanocitos a través de exosomas los que comunican el estrés al sistema inmunitario innato, especialmente a las células dendríticas que presentan antígenos a los linfocitos T. En los pacientes con vitíligo, se observa un aumento de citoquinas proinflamatorias típicas de la inmunidad innata, IL-1α, IL-1β, IL-6, IL-8, IL-12, IL-15 y TNF-α, tanto en el suero como en la piel2,3.
Patrones moleculares asociados al dañoEl fenómeno de Koebner es considerado un desencadenante inicial del vitíligo. Se han intentado identificar factores liberados durante la respuesta al estrés y el daño de los melanocitos en esta enfermedad. Los patrones moleculares asociados al daño (DAMP) son de particular interés, ya que pueden desencadenar la respuesta inflamatoria observada en la etapa activa del vitíligo, existiendo una serie de proteínas asociadas a DAMP en el vitíligo4.
El DAMP con mayor evidencia de asociación con vitíligo es la proteína de estrés calórico 70 (HSP70), que pertenece a la familia de las chaperonas intracelulares cuya función es prevenir el plegamiento incorrecto de proteínas. La sobreexpresión de HSP70 en las lesiones con vitíligo activo indica un posible papel de esta proteína en la respuesta inmune y la inflamación asociada con la enfermedad. Además, HSP70 podría considerarse un marcador potencial para evaluar la actividad de la enfermedad3,5,6.
Por otro lado, la proteína MxA (proteína 1 de resistencia al mixovirus humano), inducible por IFN-α, muestra una fuerte expresión en la piel perilesional activa del vitíligo, lo que sugiere un efecto contribuyente del IFN-α en la progresión de la enfermedad7. Asimismo, S100B es otro DAMP liberado por los melanocitos dañados, cuyos niveles están aumentados en el vitíligo activo y pueden estimular respuestas inflamatorias8. También, las proteínas del grupo de alta movilidad (HMGB1), pueden inducir la producción de ligandos de quimiocinas, como CXCL1 o IL-8 por los queratinocitos, actuando en el reclutamiento de células inmunitarias9.
La calreticulina (CRT) es otra de las moléculas más estudiadas en vitíligo, ya que induce apoptosis de los melanocitos y la liberación de productos de degradación de membrana importantes para la inmunogenicidad10.
Deficiencia en la adhesión de melanocitosVarios grupos de investigación han demostrado que los melanocitos en el vitíligo presentan propiedades adhesivas reducidas. Así, se han observado niveles alterados de expresión de E-cadherina en melanocitos de la piel afectada por vitíligo antes de que se desarrollen las despigmentaciones. La deficiente expresión de E-cadherina provoca la pérdida de adhesión de los melanocitos epidérmicos durante situaciones de estrés oxidativo o mecánico11.
Por otro lado, se ha identificado un papel relevante de las integrinas y la proteína Melanoma Inhibitory Activity (MIA). Las integrinas están involucradas en la interacción de melanocitos con la matriz extracelular, y su disfunción puede contribuir a la pérdida de melanocitos. Por otro lado, la proteína MIA, originalmente estudiada en el melanoma, ha mostrado un efecto proapoptótico sobre los melanocitos, favoreciendo su desaparición en las áreas afectadas por el vitíligo. Estos hallazgos refuerzan la idea de que la adhesión celular desempeña un papel crucial en la patogénesis de la enfermedad12.
InflamasomasLos inflamasomas son complejos multiproteicos que al activarse desencadenan una cascada de eventos que resultan en la activación de una enzima llamada caspasa-1. La activación de los inflamasomas podría desempeñar un papel crucial en la inflamación y destrucción de los melanocitos en el vitíligo no segmentario. Se ha encontrado una activación de inflamasomas en las lesiones de vitíligo, asociada a una mayor expresión de citoquinas inflamatorias como IL-1β y IL-18. Por lo tanto, la inhibición de IL-1β puede ser vista como un objetivo terapéutico potencial en vitíligo.
Los polimorfismos en IL-1β y en el inflamasoma del receptor similar a NOD 1 (NLRP1) están asociados con un mayor riesgo de desarrollar vitíligo. Al reconocer los DAMP, este receptor activa el inflamasoma, que a través de la vía de caspasa-1 induce el procesamiento de pro-IL1β en IL-1β activa. Aunque la conexión específica entre NRLP1 y el vitíligo no está completamente definida, es probable que los inflamasomas, incluido NRLP1, jueguen un papel importante en los procesos inflamatorios y de estrés celular que caracterizan esta enfermedad13,14.
Vía del interferón tipo 1 (IFN tipo 1)La vía del IFN-1 es un vínculo temprano y transitorio en la progresión de la enfermedad y marca el punto de unión entre la respuesta inmunitaria innata y adaptativa. El vitíligo está asociado con la activación del interferón tipo 1. Variantes del gen de la helicasa inducida por IFN con el dominio C 1 (IFIH1), están relacionadas con protección frente el vitíligo al inhibir la producción de IFN-α. Este interferón induce quimiocinas que reclutan células T autorreactivas como CXCL10 y CXCL16. Las células dendríticas plasmocitoides (pDC) son responsables de la producción de IFN-α en la piel con vitíligo, estimuladas por HSP703,6,15.
Inmunidad adaptativaEl daño a los melanocitos causado por el estrés oxidativo activa la inmunidad innata, con la consiguiente secreción de citoquinas y presentación de antígenos. Esto desencadena la activación del sistema inmunitario adaptativo, donde los linfocitos T autorreactivos exacerban el daño a los melanocitos en la piel afectada por el vitíligo, a través de la secreción de citoquinas proinflamatorias (fig. 1)3,4,16,17.

Mecanismos etiopatogénicos del vitíligo. Esta figura resume todas las claves importantes de cómo se desencadena una cascada molecular que comienza con una mayor susceptibilidad de los melanocitos al estrés oxidativo, que, en presencia de una base genética susceptible, da lugar a la activación del sistema inmunitario. Las células T CD8+ producen varias citoquinas como el IFN-γ. La unión del IFN-γ a su receptor activa la vía JAK-STAT y provoca la secreción de CXCL9 y CXCL10.
CCL5: ligando de quimiocina 5; CXCL9: ligando de quimiocina CXC 9; CXCL10: ligando de quimiocina CXC 10; CXCL12: ligando de quimiocina CXC 12; CXCL16: ligando de quimiocina CXC 16; CXCR3: receptor de quimioquina tipo 3; DAMP: patrón molecular asociado a daños; IFN-γ: interferón-γ; JAK: Janus quinasa; MAPK: proteína quinasa activada por mitógenos; NF-kB: factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas; NLRP1: inflamasoma del receptor similar a NOD 1; TNF-α: factor de necrosis tumoral alfa; ROS: especies reactivas del oxígeno; STAT1: transductor de señales y activador de la transcripción 1.
Los linfocitos T citotóxicos (CD8+) son las principales células inmunitarias implicadas en la patogénesis de la enfermedad, principalmente en fases iniciales o activas. El mecanismo de acción se basa en la producción de citoquinas inflamatorias como TNF-α e IFN-γ, además de la liberación de granzimas y perforinas. El IFN-γ aumenta localmente los niveles de CXCL9 y CXCL10 en la piel y el suero de los pacientes con vitíligo, lo que a su vez atrae células T patógenas que expresan el receptor CXCR3A18. En la piel perilesional se ha observado infiltrado linfocitario predominante de células T CD8+, correlacionando su actividad de la enfermedad3.
En los pacientes con vitíligo, los linfocitos T CD8+ reconocen antígenos específicos de los melanocitos (MelanA, tirosinasa, gp100, proteínas relacionadas con la tirosinasa 1 y 2, Mart-1). Estos antígenos se encuentran en cantidades significativamente mayores en la sangre periférica de los pacientes en comparación con los individuos sanos. Este fenómeno se considera la principal vía de destrucción de melanocitos en el vitíligo, aunque la presencia de células T específicas de melanocitos no es suficiente, ya que también se han encontrado en personas sanas, mostrando un fenotipo anérgico19,20.
Células T reguladorasEl desequilibrio entre la señalización proinflamatoria y antiinflamatoria es crucial en la patogénesis del vitíligo. El número de células T reguladoras (Tregs), capaces de atenuar la respuesta inmunitaria, están disminuidas en los pacientes. Además, su capacidad supresora también está comprometida21.
Se han explorado las diferencias en la respuesta inmune en los pacientes con vitíligo no segmentario y sanos, examinando los niveles de Tregs y la expresión del receptor LRP1 en monocitos. Los niveles de Tregs y LRP1/CD91 antes y después del tratamiento en los pacientes con vitíligo, comparados con individuos sanos muestran niveles superiores en los pacientes con vitíligo demostrando que las anormalidades en la respuesta inmune contribuyen a la patogénesis del vitíligo22.
Citoquinas y quimioquinasLas citoquinas, especialmente IFN-γ y TNF-α, juegan un papel crucial en la pérdida de melanocitos y la progresión del vitíligo. IFN-γ activa la vía JAK/STAT y TNF-α activa las vías MAPK y NF-kB, ambas contribuyendo a la inflamación y al daño celular. IFN-γ induce la producción de quimioquinas CXCL9 y CXCL10, que amplifican la inflamación y el reclutamiento de células inmunitarias. En modelos murinos, la inhibición de CXCL10 previno la enfermedad y promovió la repigmentación. CXCL12 y CCL5 también juegan un importante papel en el reclutamiento de células inmunitarias en la piel afectada, y los niveles de CXCL12 en el suero se asocian con la actividad de la enfermedad23. Además, el estrés oxidativo induce la secreción de quimioquinas como CXCL1624.
Otras citocinas elevadas en vitíligo incluyen IL-1β, IL-2, IL-17, IL-22, IL-23 e IL-33. Sin embargo, las terapias dirigidas contra estas moléculas han mostrado resultados contradictorios, lo que subraya la necesidad de seguir investigando para encontrar tratamientos efectivos25–27.
AutoinmunidadLa evidencia genética respalda la hipótesis autoinmune como el principal mecanismo del vitíligo, con aproximadamente el 85% de los genes de susceptibilidad implicados en la inmunidad innata, adaptativa y apoptosis4,28. Esta teoría también se refuerza con la relación del vitíligo con otros trastornos autoinmunes, la presencia de anticuerpos específicos en los pacientes y el uso de terapias inmunomoduladoras4,28.
Además, se ha observado que los tratamientos con inhibidores de puntos de control inmunitario (checkpoint inhibitors) en los pacientes con cáncer pueden inducir el desarrollo de vitíligo, lo que sugiere un vínculo entre la autoinmunidad y esta enfermedad29.
Las enfermedades autoinmunitarias son la principal comorbilidad asociada al vitíligo, incluyendo trastornos tiroideos, anemia perniciosa, alopecia areata, enfermedades del tejido conectivo, entre otras30,31.
Zombie cells, células senescentes en vitíligoLa senescencia celular se induce en respuesta a estresantes celulares. Estas células permanecen metabólicamente activas y secretan una serie de proteínas y factores bioactivos. Recientemente se ha aceptado que la presencia de células senescentes contribuye a la progresión de diversas enfermedades, entre ellas el vitíligo. Así, se encontró que un subconjunto significativo de melanocitos en las lesiones de vitíligo mostraba positividad para p16INK4A, en comparación con la piel no afectada. Este hallazgo sugiere que p16INK4A podría estar involucrado en la regulación de la función de los melanocitos en vitíligo no segmentario, posiblemente como respuesta a factores estresantes oxidativos o inflamatorios en el microambiente de la piel32.
Inmunidad entrenada y tolerancia inmuneUn aumento a largo plazo en la función de memoria innata, descrita como inmunidad entrenada después de la vacunación y en otras enfermedades inflamatorias, podría jugar un papel como potenciador y desencadenante continuo en la patogénesis del vitíligo. Tras la exposición a ciertos estímulos, el sistema inmunitario innato es capaz de mostrar una respuesta inmunológica mejorada a un segundo estímulo, lo que indica una función de memoria del sistema inmunitario innato, concepto conocido como inmunidad entrenada. La inmunidad entrenada está regulada por la reprogramación epigenética, que incluye modificaciones químicas de histonas, así como en la accesibilidad de la cromatina que causan cambios sostenidos en la transcripción de genes específicos33.
Células T memoria residentesLa recurrencia del vitíligo en lugares previamente afectados demuestra una respuesta de memoria implicada en la etiopatogenia de esta enfermedad. Este riesgo de recaída podría deberse a la persistencia de células T de memoria residentes en el tejido (TRM), cuyo mantenimiento y función son promovidos por la IL-15. Estas células TRM presentan niveles altos de CD122, la subunidad del receptor de IL-1534–37. Este anticuerpo ha demostrado la reversión de la enfermedad en ratones, por lo que podría ser una diana terapéutica prometedora38. Recientemente se ha demostrado que la supervivencia de las TRM depende de la captación y metabolismo de ácidos grasos exógenos, lo que sugiere que un posible tratamiento podría ser la regulación del metabolismo lipídico, enfocado a la eliminación de las TRM en los tejidos periféricos39.
Anomalías intrínsecas en los melanocitos y alteraciones epidérmicas y dérmicas en la piel con vitíligoVarios estudios in vitro e in vivo han demostrado la presencia de anomalías intrínsecas en los melanocitos del vitíligo caracterizados por presentar una mayor susceptibilidad a los agentes prooxidantes. En la epidermis de las lesiones con vitíligo se ha detectado una marcada disminución en la expresión de c-kit, el receptor del factor de células madre y el factor de transcripción MITF (factor de transcripción asociado con microftalmia), junto con una reducción en el receptor de endotelina B40. También se han observado una marcada reducción en la expresión del microARN-211, asociada a una menor tasa de consumo de oxígeno, complejos mitocondriales aberrantes, alteraciones en el metabolismo lipídico y un aumento de las especies reactivas de oxígeno. Este microARN-211 podría servir en el futuro como un biomarcador para evaluar la respuesta terapéutica en esta enfermedad41.
El papel del estrés del retículo endoplasmático (RE) en la patogénesis del vitíligo ha sido examinado debido a su conexión con la acumulación de proteínas mal plegadas, lo que activa la respuesta de proteínas no plegadas (UPR). Esta respuesta intenta restaurar la homeostasis celular, pero si falla, puede contribuir al desarrollo de enfermedades autoinmunes. En el vitíligo, el estrés del RE podría vincular el estrés oxidativo con la autoinmunidad. El estrés oxidativo altera el potencial redox celular, extendiéndose al RE y causando acumulación de proteínas mal plegadas. La UPR, crucial para la inmunidad innata y adaptativa, se activa en esta situación, sugiriendo su papel en la regulación y mantenimiento de la respuesta inmune en el vitíligo42.
Otro estudió reciente analizó el papel del metabolismo energético mitocondrial en melanocitos de los pacientes con vitíligo, encontrando una producción reducida de ATP, aumento en la fuga de protones, alteración en la expresión de enzimas glucolíticas e hiperactividad del eje PGC1α. Además, se demostró que la estabilización farmacológica de la cardiolipina, un lípido clave en la membrana mitocondrial, puede revertir la disfunción energética observada en los melanocitos de vitíligo. Esto sugiere que la manipulación de la cardiolipina podría ser una nueva estrategia terapéutica prometedora para tratar el vitíligo43.
La piel con vitíligo muestra cambios morfológicos en el epitelio y en la dermis superior. Histológicamente, se observa una menor pigmentación en la capa basal, acompañada de una ausencia de melanocitos en las lesiones, lo cual puede confirmarse mediante técnicas de inmunohistoquímica específicas, como la tinción para proteínas HMB-45 o S100. En ocasiones, se observa un patrón de dermatitis liquenoide asociado, con linfocitos T CD3+, CD8+, más evidente en piel perilesional. Por otro lado, las células de Langerhans están aumentadas en la epidermis, la membrana basal está engrosada y se observan fenómenos de vacuolización citoplasmática44,45. Cabe destacar que la proteína Dickkopf 1 (DKK1), implicada en la reducción de la melanogénesis, está sobreexpresada en fibroblastos en piel lesional46. También hay una mayor expresión de fibronectina y elastina reducida en dermis lesional, sin observarse cambios en las fibras de colágeno47,48.
Alteraciones genéticas implicadas en la etiopatogenia del vitíligoEl vitíligo es una enfermedad compleja con un componente genético significativo, estimado entre un 75 y un 83%. Esta teoría involucra múltiples genes, la mayoría de los cuales corresponden al sistema inmunológico, con un menor número de ellos implicando a los melanocitos. Existe una notable superposición entre los genes implicados en el vitíligo y aquellos que se han asociado a trastornos autoinmunitarios. Los familiares de primer grado de los pacientes con vitíligo presentan un riesgo del 6-8% de desarrollar la enfermedad, y la tasa de concordancia entre gemelos monocigóticos es aproximadamente del 23%3,4,28.
Se han realizado 5 estudios de asociación del genoma completo (GWAS) en el vitíligo en poblaciones europeas y asiáticas, identificando al menos 54 loci de susceptibilidad al vitíligo49,50.
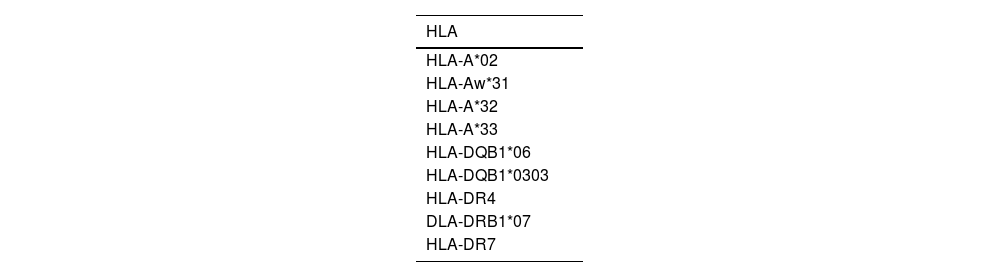
Región del antígeno leucocitario humanoVarios genes dentro de la región del antígeno leucocitario humano (HLA), específicamente en las regiones de clase I y II, están asociados con el vitíligo. Estos estudios incluyen análisis de asociaciones de GWAS y estudios de ligamiento en familias afectadas. La patogénesis del vitíligo ha sido relacionada con el gen XBP1, un factor de transcripción que juega un papel crucial en la respuesta al estrés del retículo endoplásmico y regula la expresión de genes HLA de clase II51. En población china se han identificado HLA-DQB1 y HLA-B como factores de riesgo asociados, en europeas 3 loci específicos y uno en asiáticos (vinculados respectivamente a FOXD3 y PDGFRA)52–54. Por otro lado, hay estudios que correlacionan HLA-A*09 y HLA-Aw*19 con un menor riesgo para la enfermedad55 (tabla 1).
Genes HLA asociados con un mayor riesgo de desarrollar vitíligo. Estos estudios incluyen análisis de asociaciones de genoma completo (GWAS) y estudios de ligamiento en familias afectadas51–60
| HLA |
|---|
| HLA-A*02 |
| HLA-Aw*31 |
| HLA-A*32 |
| HLA-A*33 |
| HLA-DQB1*06 |
| HLA-DQB1*0303 |
| HLA-DR4 |
| DLA-DRB1*07 |
| HLA-DR7 |
HLA: antígeno leucocitario humano.
También se han identificado distintos genes vinculados a la susceptibilidad al vitíligo relacionados con la inmunorregulación de la inmunidad innata y adaptativa (tablas 2 y 3). Las variantes del gen PTPN22 se han vinculado con varias enfermedades autoinmunes, como la artritis reumatoide o el lupus eritematoso sistémico, lo que lo convierte en un gen con un papel relevante en el contexto de enfermedades autoinmunes61. También se han vinculado, los genes IKZF4 y FOXP3 que ejercen un papel de silenciamiento génico en células Tregs62.
Principales genes involucrados en la etiopatogenia del vitíligo
| Gen | Cromosoma y región | Función |
|---|---|---|
| XBP151 | Cromosoma 22q12 | Factor de transcripción que regula la expresión de HLA II. Involucrado en la respuesta celular al estrés |
| FOXD353 | Cromosoma 1p31.3 - p32.2 | Factor de transcripción implicado en la regulación de la diferenciación celular y la respuesta inmunológica |
| PDGFRA52 | Cromosoma 4q13-q21, | Involucrado en la proliferación celular y angiogénesis |
| NLRP164 | Cromosoma 17p13 | Detección de patógenos, formación del inflamasoma, activación de la caspasa-1, secreción de citoquinas e inducción de piroptosis |
| PTPN2261 | Cromosoma 1p13.2 | Codifica una fosfatasa de tirosina involucrada en la señalización de células T, relacionada con varias enfermedades autoinmunes |
| IKZF462 | Cromosoma 12q13.2 | Silenciamiento génico mediado por FOXP3 en células Tregs |
| FOXP362 | Cromosoma X | Silenciamiento génico en células Tregs |
| DDR165 | Cromosoma 6p21.3 | Codifica un receptor tirosina quinasa, involucrada en la adhesión de melanocitos a la membrana basal |
| VEGF66 | Cromosoma 6p21.1 | Regulador de la angiogénesis involucrado en enfermedades crónicas y neoplasias. |
HLA: antígeno leucocitario humano.
Genes inmunorreguladores implicados en el desarrollo, la activación, la señalización de células T y la respuesta inmune innata vinculados al vitíligo4,28,49,50
| Desarrollo de células T | Señalización del receptor de células T | Activación de células T | Respuesta inmunidad innata | Receptores de quimioquinas y citoquinas |
|---|---|---|---|---|
| CD44 | SLA | BTNL2 | IFIH1 | CXCR5 |
| CD80 | PTPN22 | FOXP3 | TICAM1 | CCR6 |
| UBASH3A | IKZF4 | SH2B3 | ||
| CLNK | IL2RA | |||
| CTLA4 |
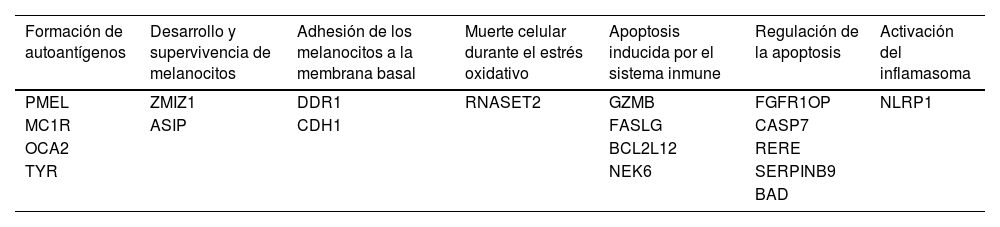
Varios genes relacionados con la función y supervivencia de los melanocitos han sido implicados en el vitíligo (tabla 4). El gen TYR, implicado en biosíntesis de melanina en los melanocitos ha sido asociado significativamente en la población europea con vitíligo63. Los roles de muchos loci de susceptibilidad continúan siendo desconocidos, lo que destaca la falta de compresión en la etiopatogenia del vitíligo que continúa siendo un desafío en la actualidad. Estos conocimientos sobre la genética ponen en manifiesto la naturaleza multifactorial del vitíligo con un patrón de herencia poligénico, involucrando una interacción compleja entre la disfunción del sistema inmunitario y la disfunción de los melanocitos.
Genes relacionados con la función y supervivencia de melanocitos4,28,49,50
| Formación de autoantígenos | Desarrollo y supervivencia de melanocitos | Adhesión de los melanocitos a la membrana basal | Muerte celular durante el estrés oxidativo | Apoptosis inducida por el sistema inmune | Regulación de la apoptosis | Activación del inflamasoma |
|---|---|---|---|---|---|---|
| PMEL | ZMIZ1 | DDR1 | RNASET2 | GZMB | FGFR1OP | NLRP1 |
| MC1R | ASIP | CDH1 | FASLG | CASP7 | ||
| OCA2 | BCL2L12 | RERE | ||||
| TYR | NEK6 | SERPINB9 | ||||
| BAD |
El estrés oxidativo desempeña un papel fundamental en la patogénesis del vitíligo, contribuyendo al daño y la destrucción de los melanocitos. El estrés oxidativo se refiere al desequilibrio entre la producción de especies reactivas de oxígeno (ROS) y la capacidad antioxidante del cuerpo para neutralizarlas. En los pacientes con vitíligo, se ha observado una acumulación excesiva de ROS en los melanocitos que pueden dañar componentes celulares cruciales, incluyendo lípidos, proteínas y ADN, lo que lleva a su disfunción y muerte. El daño celular causado por el estrés oxidativo puede liberar autoantígenos y activar una respuesta autoinmune, contribuyendo a la destrucción de los melanocitos4,28,67,68. Estudios han demostrado niveles elevados de biomarcadores de estrés oxidativo, como el malondialdehído (MDA) y la disminución de antioxidantes, como el superóxido dismutasa (SOD) y la catalasa, tanto en la piel afectada como en la sangre de los pacientes. Además, determinadas variantes en genes relacionados con la respuesta antioxidante, como el gen de la catalasa (CAT) y el gen del glutatión peroxidasa (GPX), se han asociado con una mayor susceptibilidad al vitíligo67,69,70.
El vitíligo también se ha asociado con un reciclaje defectuoso de la tetrahidrobiopterina (6BH4), esencial para la formación de melanina, lo que lleva a un exceso de 7BH4 y la inhibición de la fenilalanina hidroxilasa (PAH). Esto produce peróxido de hidrógeno (H2O2), que disminuye la actividad de la enzima dihidropteridina reductasa (DHPR)71,72. Además, el H2O2 afecta negativamente la acetilcolinesterasa (AchE) y la xantina oxidasa (XO)73,74.
Los mecanismos de reparación y la activación del sistema inmunológico en los pacientes con vitíligo podrían reducir el riesgo de cáncer de piel. Esto se atribuye a la activación inmunológica que es capaz de identificar y eliminar células dañadas, incluyendo aquellas con mutaciones potencialmente precancerosas. Además, el estrés oxidativo y la autoinmunidad en el vitíligo generan un microambiente menos favorable para el desarrollo de cáncer de piel, incluyendo el melanoma75.
Agentes ambientales como activadores de la enfermedadExisten factores ambientales que influyen en la aparición o empeoramiento del vitíligo en las personas susceptibles (tabla 5). El trauma físico, conocido como el fenómeno de Koebner, puede inducir la aparición de manchas de vitíligo en áreas de fricción repetida, heridas, cicatrices y lesiones preexistentes de eccema de contacto o psoriasis76. La exposición a químicos, como derivados fenólicos, genera un efecto melanotóxico en el vitíligo, al inducir estrés oxidativo y causar daño directo en los melanocitos. Además, tatuajes, vacunas y ciertos cosméticos, también puede precipitar el vitíligo al alterar los melanocitos. El calor, derivado de procedimientos como la depilación láser o la luz pulsada, puede dañar la piel y desencadenar la despigmentación77,78. Por otro lado, el tabaquismo, con sus efectos perjudiciales sobre la salud de la piel y el sistema inmunológico, puede exacerbar la enfermedad79. Además, numerosos medicamentos, incluyendo algunos inmunomoduladores y agentes biológicos, pueden inducir o empeorar el vitíligo80–83. Las infecciones virales, como el VHC, VIH, virus varicela-zóster (VVZ) y COVID-19, así como la exposición a alérgenos como ácaros del polvo, también se han asociado con brotes de vitíligo84–86. Finalmente, el estrés psicológico es un factor conocido que puede agravar la condición, posiblemente debido a su impacto sobre el sistema inmunológico87. Sin embargo, no hay estudios que comprueben una asociación directa entre el vitíligo y la contaminación, dietas específicas o consumo de alcohol88–90.
Agentes ambientales como activadores del vitíligo76–86,88–91
| Agentes ambientales | |
|---|---|
| Químicos | Derivados fenólicos (monobenzona, rododendrol), tatuajes, vacunas, tintes cosméticos (parafenilendiamina) |
| Calor | Depilación láser, luz pulsada |
| Trauma | Fenómeno de Koebner en zonas de trauma repetido, fricción, heridas, uso de mascarillas, lesiones residuales de eccema de contacto, psoriasis, etc. |
| Medicamentos | Paracetamol (acetanominofen), fármacos usados en los pacientes hematológicos (mogamulizumab, ITK), fármacos usados en los pacientes dermatológicos (anti-TNF, dupilumab, secukinumab, imiquimod, difenciprona), fármacos usados en los pacientes neurológicos (alemtuzumab, carbamazepina, tolcapona, levodopa), antibióticos (cloroquina, clofazimina), betabloqueantes, fármacos anti-PD1 (pembrolizumab, nivolumab), fármacos anti-CTLA-4 (ipilimumab) |
| Infecciones | VIH, VHC, herpesvirus (virus varicela-zóster, virus herpes simple), COVID-19 |
| Tabaquismo | |
| Estrés psicológico |
ITK: inhibidores de tirosina cinasa; VHC: virus de la hepatitis C; VIH: virus de inmunodeficiencia humana.
La hipótesis neural sugiere que el vitíligo segmentario está relacionado con la disfunción del sistema nervioso. Se postula que los neurotransmisores o neuroquímicos liberados por las terminaciones nerviosas, en concreto la sustancia P, pueden ser tóxicos para los melanocitos, causando su destrucción en áreas específicas. La distribución segmentaria de las manchas de vitíligo corresponde a la inervación de ciertos nervios, apoyando esta hipótesis92,93. Un estudio que analiza la presencia del VVZ en piel con vitíligo segmentario sugiere una posible relación entre ambos. Los resultados indican la presencia de partículas virales y cambios relacionados en la piel afectada, sugiriendo que el VVZ podría contribuir al desarrollo o progresión del vitíligo86.
En ocasiones el vitíligo segmentario no sigue una distribución dermatómica, lo que sugiere que los mecanismos neuronales no son la única causa atribuible. Por ello, el mosaicismo genético es en la actualidad la teoría más aceptada para explicar esta condición, aunque aún no se ha confirmado genéticamente94.
Nuevas vías patogénicas y posibles nuevas dianas terapéuticasLa patogénesis del vitíligo es multifactorial, involucrando una interacción compleja entre la predisposición genética y diversos factores ambientales, estrés oxidativo y respuestas autoinmunes. El principal desafío en la creación de modelos fisiopatológicos es integrar estas diversas teorías y elementos.
El descubrimiento de nuevas vías patogénicas plantea posibles nuevas dianas terapéuticas sobre las que actuar. Las estrategias terapéuticas emergentes incluyen la reducción de especies reactivas de oxígeno (ROS), la inhibición de la señalización de IFN-γ e IL-15 mediante la vía JAK-STAT, y el uso de anti-IL15 y anti-CD122. Además, la reducción de células T residentes de memoria y la intervención en la vía NRLP1 actuando sobre el inflamasoma también son enfoques prometedores para tratar esta enfermedad.
AutoríaTodos los autores contribuyeron a la preparación del manuscrito y modificado críticamente, y aprobaron la versión presentada.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



