
























Vascular malformations are inborn errors of vascular embryogenesis present at birth that should be diagnosed in childhood and, when necessary, treated to prevent later complications. The current trend is to classify these lesions according to flow characteristics and the predominant type of vascular channel affected.
Given the complexity, and in many cases, the rarity, of vascular malformations, they should be managed by multidisciplinary teams at vascular anomalies centers.
Furthermore, because the association between vascular malformations and certain syndromes is becoming increasingly recognized, a better understanding of these lesions will help to improve overall patient management in this setting.
Las malformaciones vasculares (MV) son errores innatos en el desarrollo embriológico de los vasos sanguíneos que están presentes siempre desde el nacimiento. Por ello deben diagnosticarse en la infancia y, en los casos en que sea necesario, tratarlas para evitar complicaciones posteriores. La tendencia actual es a clasificar estas lesiones en cuanto al flujo que presentan y en cuanto al tipo de vaso predominante en las mismas.
Dada la complejidad que presentan estas lesiones, y en muchos casos su poca frecuencia, deben abordarse desde un punto de vista multidisciplinario en centros de anomalías vasculares.
Además, la asociación de malformaciones vasculares con cuadros sindrómicos está cada vez mejor definida, lo que hace necesario su conocimiento para un mejor abordaje de estos enfermos.
Vascular anomalies are a group of diseases that have caused confusion throughout the history of medicine The main obstacle to the advancement of knowledge in this field has been a consistent lack of standardized nomenclature.
A major breakthrough in this regard was the publication of a classification system based on clinical, radiologic, hemodynamic, and histopathologic criteria by Mulliken and Glowacki1 in 1982. Their work formed the groundwork for what is now the most widely accepted classification system for vascular anomalies: the International Society for the Study of Vascular Anomalies (ISSVA) classification. Thanks to the development of this standardized system, the past 2 decades have seen spectacular advances in our knowledge of the genetic determinants, pathophysiology, diagnosis, and treatment of vascular anomalies.2
Definition, Classification, and ImportanceThe ISSVA classification divides benign vascular lesions into 2 groups: vascular tumors (the most common of which is infantile hemangioma) and vascular malformations.
Vascular malformations arise from inborn errors of vascular embryogenesis. They are always present at birth but they can sometimes present or cause problems later. They usually develop gradually, but their growth is proportionally faster than that of the patient, with peak growth occurring during puberty.
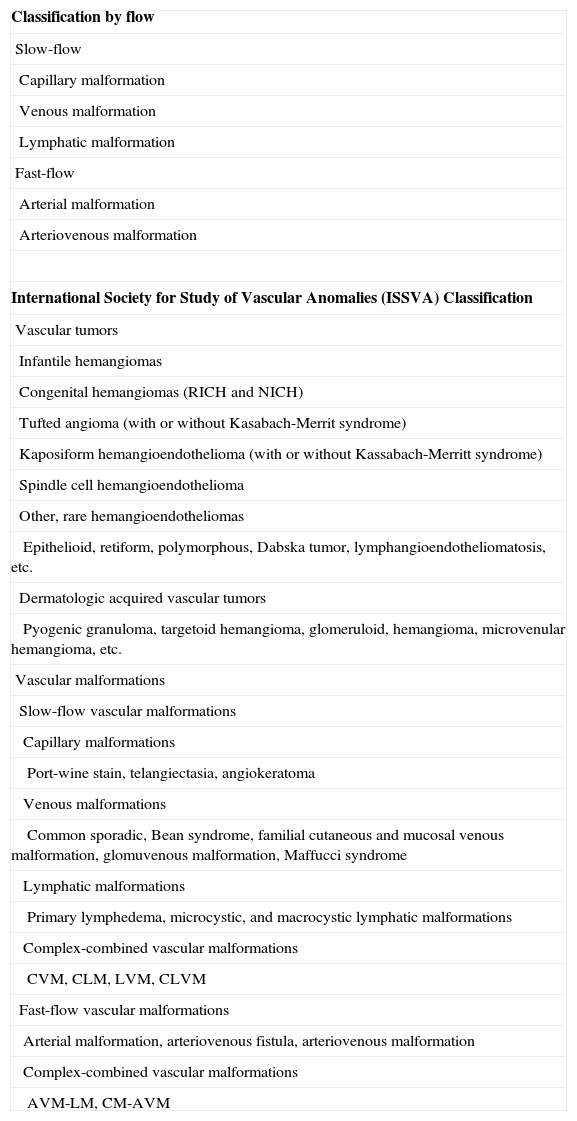
The ISSVA, founded in Budapest in 1992, classifies vascular malformations according to the predominant type of vascular channel affected (Table 1). However, it also divides them according to flow3 (Table 1) because the hemodynamic characteristics are very important from a functional perspective and have both diagnostic and therapeutic implications.
Classification of Vascular Malformations by Flow.
| Classification by flow |
| Slow-flow |
| Capillary malformation |
| Venous malformation |
| Lymphatic malformation |
| Fast-flow |
| Arterial malformation |
| Arteriovenous malformation |
| International Society for Study of Vascular Anomalies (ISSVA) Classification |
| Vascular tumors |
| Infantile hemangiomas |
| Congenital hemangiomas (RICH and NICH) |
| Tufted angioma (with or without Kasabach-Merrit syndrome) |
| Kaposiform hemangioendothelioma (with or without Kassabach-Merritt syndrome) |
| Spindle cell hemangioendothelioma |
| Other, rare hemangioendotheliomas |
| Epithelioid, retiform, polymorphous, Dabska tumor, lymphangioendotheliomatosis, etc. |
| Dermatologic acquired vascular tumors |
| Pyogenic granuloma, targetoid hemangioma, glomeruloid, hemangioma, microvenular hemangioma, etc. |
| Vascular malformations |
| Slow-flow vascular malformations |
| Capillary malformations |
| Port-wine stain, telangiectasia, angiokeratoma |
| Venous malformations |
| Common sporadic, Bean syndrome, familial cutaneous and mucosal venous malformation, glomuvenous malformation, Maffucci syndrome |
| Lymphatic malformations |
| Primary lymphedema, microcystic, and macrocystic lymphatic malformations |
| Complex-combined vascular malformations |
| CVM, CLM, LVM, CLVM |
| Fast-flow vascular malformations |
| Arterial malformation, arteriovenous fistula, arteriovenous malformation |
| Complex-combined vascular malformations |
| AVM-LM, CM-AVM |
Abbreviations: A, arterial; C, capillary; L, lymphatic; M, malformation; NICH, noninvoluting congenital hemangioma; RICH, rapidly involuting congenital hemangioma; V, venous.
The earliest studies of vascular malformations reported that these lesions were not characterized by cell proliferation but were rather dysplastic vessels with normal endothelial turnover. This theory, however, has now been brought into question, particularly with regard to arteriovenous malformations, as they sometimes exhibit tumor-like growth and induce the proliferation of endothelial cells.4
Familiarity with vascular malformations is very important as they can occur alone or as part of more complex syndromes that affect other organs. The syndromes associated with vascular malformations in childhood are addressed in the second part of this article.
Capillary MalformationsCapillary malformations are probably the most heterogeneous group within the ISSVA vascular malformation classification, although this heterogeneity has been criticized by some authors.5
They are defined as malformations in which the predominant vessels are small, slow-flow vessels (i.e., arterioles and postcapillary venules).
Excluding the salmon patch, capillary malformations occur in approximately 0.3% of the general population and have no predilection for sex. While they are generally sporadic, there have been reports of familial cases.
Salmon PatchThe salmon patch is the most common capillary malformation, occurring in up to 50% of all neonates.6,7 It is a pinkish macule that typically affects the back of the neck (stork bite) or the glabella (angel's kiss), although it can affect the eyelids, the nose, the upper lip, and the sacral area (Fig. 1). The salmon patch tends to occur as a solitary lesion and generally disappears spontaneously but it can last for a lifetime. It is important to monitor salmon patches during the first months of life to ensure that they are not precursor lesions of hemangiomas. Salmon patches with a darker hue may be confused with port-wine stains. In such cases, the diagnosis is determined by the clinical course of the lesion.

The salmon patch responds very well to vascular-selective laser therapy (e.g., pulsed-dye laser therapy), but treatment is generally not considered necessary.
Salmon patches are rarely biopsied because of their benign nature and the fact that they usually resolve spontaneously in a matter of months. Biopsy findings, however, are of great interest because these patches are histologically similar to port-wine stains (discussed below), which never disappear. A better understanding of the pathophysiologic mechanisms underlying the involution of salmon patches could help to guide the treatment of capillary malformations that do not resolve spontaneously.
Port-Wine StainPort-wine stains are present at birth and continue to grow during the first months of life, when they reach their maximum size. Some of these lesions temporarily become lighter in color but the mechanisms responsible for this color loss are poorly understood.
Port-wine stains present as pink to purple macules and can occur on any part of the body although the most common location is the face (Fig. 2). They sometimes form part of more complex syndromes (Table 2).
 on the chin.")
Syndromes Associated With Port-Wine Stain Capillary Malformation.
| Sturge-Weber syndrome |
| Klippel-Trenaunay-Webner syndrome |
| Parkes-Weber syndrome |
| Phacomatosis pigmentovascularis |
| Proteus syndrome |
| Cobb syndrome |
| Bannayan-Riley-Ruvalcaba syndrome |
| Servelle-Martorell syndrome |
| Beckwith-Wiedemann syndrome |
| Roberts (pseudothalidomide) syndrome |
| Thrombocytopenia with absent radii (TAR) syndrome |
| Von Hippel-Lindau disease |
| Rubinstein-Taybi syndrome |
| Coats syndrome |
| CLAPOa syndrome |
| CLOVESb syndrome |
| RASA1: capillary malformation-arteriovenous malformation syndrome |
| Angiomyolipomas and port-wine stain |
| Macrocephaly-capillary malformation (cutis marmorata telangiectatica congenita) |
Histologic examination shows increased vascularity in the papillary and reticular dermis and vascular ectasia due to a lack of nerve fibers in the affected vessels.
Port-wine stains generally become deeper, thicker, and darker in color over time, mainly as a result of progressive vascular dilatation. As a result of these changes, which are generally much more evident on the face, patients often develop hypertrophy and nodularity after the third or fourth decade of life. The fact that nodules are only found on the face and do not occur in childhood has led some authors to suggest that port-wine stains might actually be hamartomatous lesions.8
Diagnosis is essentially clinical,9 but it is important to remember that, particularly in childhood, port-wine stains cannot be distinguished from stable arteriovenous malformations, even with the use of flow or other imaging studies.
Depending on their location, port-wine stains may be a marker of associated malformations. Particular attention should be paid to lesions in the first branch of the trigeminal nerve (Sturge-Weber syndrome),10 in the lumbar region (spinal dysraphism),11,12 in the dorsolumbar region (Cobb syndrome), and on the lower lip (CLAPO syndrome involving capillary malformation of the lower lip [C], lymphatic malformation of the face and neck [L], asymmetry of the face and limbs [A], and partial or generalized overgrowth [O]).
Vascular laser therapy is the treatment of choice. Pulsed-dye lasers are the most widely used lasers in this setting,13,14 but satisfactory results have also been achieved with long-pulsed Nd:YAG lasers, KTP lasers, and intense pulsed light sources.15,16 Despite the advances that have been made in this area, only 25% to 30% of port-wine stains are completely eliminated by laser therapy. Improved results, however, appear to be possible with the application of cold during the delivery of energy, with the use of a 755-nm laser,17 and with dual wavelength treatment using a pulsed dye laser and an Nd:YAG laser.18 Laser therapy combined with imiquimod19 and topical angiogenesis inhibitors such as rapamycin20,21 has also been tested.
The best time to start treatment has been the subject of debate for years. It is currently thought that port-wine stains would benefit from early treatment because they become deeper and hypertrophic with time. Laser therapy is performed under general anesthesia with sedation in many centers. This can be problematic for several reasons. First, it requires the use of an operating room (which means additional health care costs), and second, the repeated use of sedation to treat a largely cosmetic problem is not without risk, although the psychologic impact of these lesions on patients and their families, cannot, of course, be overlooked.22–24 Advances in pediatric anesthesia have led to a number of alternatives, such as nitric oxide and in-office sedation, that allow the treatment of larger surface areas.
Port-wine stains may be accompanied by soft tissue hypertrophy, particularly when the extremities are affected. It is important to recognize the combination of stable hemihypertrophy associated with capillary malformations because it is a benign malformation and the relative difference present at birth remains constant throughout the patient's life. As it is sometimes difficult to distinguish this condition from other entities involving overgrowth (e.g. Klippel-Trenaunay-Webner syndrome), diagnostic errors are common.
There is also a group of acquired port-wine stains with characteristics identical to those of the congenital variant.25
Finally, in recent years, an association has been described between multiple, small port-wine stains associated with cerebral arteriovenous malformations and the RASA1 gene.26
Nevus AnemicusNevus anemicus is a nonhereditary congenital condition characterized by irregular hypopigmented macules that coalesce to form plaques and occur particularly on the chest. It is generally present at birth or develops in the first days of life. It is more common in females. Diagnosis is confirmed by applying gentle friction to the lesion and the surrounding skin and checking that the erythema produced in the healthy skin does not appear in the hypopigmented lesion27 (Fig. 3).

The condition was first described in 1906 by Van Hömer, who reported a reduction in the number and size of blood vessels, which he attributed to possible partial aplasia of the dermal vessels. It was later shown, however, that the number of vessels in the area was normal and that the pallor was due to vascular hypersensitivity to catecholamines (i.e., sustained vasoconstriction).28 When skin from a nevus anemicus is biopsied and grafted onto normal skin, it retains its pallor. It has also been shown that skin affected by nevus anemicus has reduced delayed hypersensitivity because the vessels do not respond adequately to proinflammatory cytokines.29
Nevus anemicus normally occurs in isolation in healthy individuals, but it has been described in patients with neurofibromatosis and in patients with a port-wine stain,30 some of whom had Sturge-Weber syndrome. The co-occurrence of a port-wine stain, nevus anemicus, and Mongolian spots is known as phacomatosis pigmentovascularis type IIb. Nevus anemicus has also been associated with nevus spilus, primary lymphedema, alopecia universalis, onychodystrophy, extensive vitiligo, and atopy.
The differential diagnosis of nevus anemicus should include pityriasis versicolor, vitiligo, nevus depigmentosus, and the hypopigmented macules of tuberous sclerosis and leprosy.
Nevus anemicus is not included in the ISSVA classification of capillary malformations, but it probably should be.
Nevus RoseusIn 2005, Happle31 described a variant of port-wine stain that was characterized by a pale red or even pink tone, in contrast to the darker hue of the port-wine stain (Fig. 4). By analogy with the term port-wine stain, he proposed calling this new variant rosé-wine stain, or nevus roseus. Nevus roseus, however, cannot be definitely diagnosed until adulthood as port-wine stains are sometimes pink in children. While the natural history of port-wine stains includes hypertrophy, darkening, and nodularity, nevus roseus remains unchanged for life.

The differential diagnosis should also include the salmon patch, but unlike nevus roseus and port-wine stains, which tend to be lateralized, salmon patches always affect the midline of the body.
The fact that nevus roseus occurs in association with phacomatosis pigmentovascularis type III (phacomatosis spilorosea) is the strongest evidence that it is a separate entity.
Cutis Marmorata Telangiectatica CongenitaCutis marmorata telangiectatica congenita is a rare, sporadic congenital vascular malformation first described by Van Louizen in 1922. It presents as localized or generalized erythematous-telangiectatic lesions with a reticular pattern; the lesions are almost always present at birth or develop in the first days of life32 (Fig. 5). In the largest series published to date, consisting of 85 patients, 60% of patients had localized lesions and 65% had unilateral lesions, mostly affecting the lower limbs.33 Twenty percent of patients with cutis marmorata telangiectatica congenita have capillary malformations in other locations, including port-wine stains, nevus anemicus, and angiokeratomas34; there has even been a report of cutis marmorata telangiectatica congenita initially manifesting as a port-wine stain.35 The lower limbs are the most common sites affected, and in a third of these cases, the skin in the affected area is atrophic. On the upper limbs, by contrast, hypertrophy is more common (up to 20% of cases).

Cutis marmorata telangiectatica congenita occurs in association with other conditions in 20% of patients (Table 3). These conditions include hemangioma, congenital pigmented nevus, café au lait spots, hypospadias, glaucoma, hemispheric arteriovenous malformation, syndactyly, aplasia cutis congenita, heart defects, multicystic renal disease, and congenital hypothyroidism.36
Cutis Marmorata Telangiectatica Congenita: Associations.
| Congenital hypothyroidism |
| Glaucoma |
| Hemangioma |
| Congenital pigmented nevus |
| Café au lait macules |
| Hypospadias |
| Syndactyly and aplasia cutis |
| Multicystic renal disease |
| Nevus flammeus |
| Neonatal lupus |
| Monoatrophy |
| Transient ascites and elevated human chorionic gonadotropin hormone levels |
| Elevated copper levels and increased elastolysis |
| Adams-Oliver syndrome |
| Extensive Mongolian spots: phacomatosis cesiomarmorata |
| Microcephalic osteodysplastic primordial dwarfism type II |
| Chronic autoimmune urticaria |
| Hypoplasia of right iliac and femoral veins |
It is important to note that cutis marmorata telangiectatica congenita can be confused with neonatal lupus.37,38 Depending on the patient's history, it may be necessary to perform additional tests.
Cutis marmorata telangiectatica congenita lesions generally disappear spontaneously after several months or years, but they occasionally persist throughout life. They most often occur in association with capillary malformations, and it is therefore thought that some of these anomalies might be an extension of cutis marmorata telangiectatica congenita. Indeed, several persistent cases of cutis marmorata telangiectatica congenita are in effect port-wine stains with a reticular pattern.
In the new classification of phacomatosis pigmentovascularis, cutis marmorata telangiectatica congenita is part of phacomatosis cesiomarmarota. These patients also have extensive Mongolian spots.39
Macrocephaly-capillary malformation, which is discussed in the section on associated syndromes, was originally called macrocephaly-CMTC but was renamed when it was found that the condition involved capillary malformations other than cutis marmorata telangiectatica congenita.
Phacomatosis PigmentovascularisPhacomatosis pigmentovascularis is an association between a vascular nevus and an extensive pigmentary nevus40 (Fig. 6). It was named by Ota in 1947, but it had been described earlier. The initial classification system divides phacomatosis pigmentovascularis into 5 groups according to the associated pigmentary anomaly.41 This classification was later simplified by Happle.42 The 2 systems are shown in Table 4. The etiology of phacomatosis pigmentovascularis involves the genetic mechanism of twin spotting (didymosis), a phenomenon much more widely described in animals and plants in which cells lose their genetic heterozygosity.

Classification of Phacomatosis Pigmentovascularis.
| Original Classification | |
| Type I | |
| Nevus flammeus and pigmented linear epidermal nevi | |
| Type II | |
| Nevus flammeus and Mongolian spots and/or nevus anemicus | |
| Type III | |
| Nevus flammeus and nevus spilus and/or nevus anemicus | |
| Type IV | |
| Nevus flammeus, Mongolian spots, and nevus spilus and/or nevus anemicus | |
| Type V | |
| Cutis marmorata telangiectatica congenita and Mongolian spots | |
| Unclassifiable | |
| Type A or B depending on whether or not there is systemic involvement | |
| Happle's Shortened Classification | |
| Phacomatosis cesioflammea | |
| Nevus cesius (blue spot) and nevus flammeus | |
| Phacomatosis spilorosea | |
| Nevus spilus and telangiectatic nevus of a pale-pink type (nevus roseus) | |
| Phacomatosis cesiomarmorata | |
| Nevus cesius (blue spot) and cutis marmorata telangiectatica congenita | |
| Phacomatosis pigmentovascularis, unclassifiable type | |
| Correspondence Between 2 Systems | |
| Cesioflammea | IIA, IIB |
| Spilorosea | IIIA, IIIB |
| Cesiomarmorata | VA, VB |
| Unclassifiable | IA, IVB |
| Type I from the first classification is eliminated | |
Diagnosis is mainly clinical and the disorder, in particular phacomatosis cesioflammea, has been associated with multiple anomalies (Table 5). There have also been a number of reports of an association with complex-combined vascular malformations, such as Klippel-Trenaunay-Webner syndrome.43
Associations Reported for Phacomatosis Cesioflammea.
| Skin lesions |
| Nevus anemicus, café au lait spots, alopecia universalis, congenital triangular alopecia |
| Vascular anomalies |
| Sturge-Weber syndrome, Klippel-Trenaunay syndrome, pyogenic granuloma, venous malformations |
| Neurologic disorders |
| Seizures, cortical atrophy, Arnold-Chiari type I malformation, bilateral deafness, idiopathic facial paralysis, hydrocephalus, diabetes insipidus, plexiform neurofibroma, psychomotor retardation |
| Eye alterations |
| Ocular melanosis, iris mammillations and hamartomas, glaucoma, prominent scleral vessels, chronic corneal edema, retinal pigment alterations, pigmentary retinosis |
| Miscellaneous |
| Discrepancy in limb length, scoliosis, spinal dysraphism, hemihypertrophy, syndactyly, macrocephaly, renal angiomatosis, hepatosplenomegaly, umbilical hernia, hypoplasia of leg veins, immunoglobulin A deficiency, hyperimmunoglobulin E syndrome, eczema, and premature tooth eruption. |
Telangiectasias are small, permanently dilated blood vessels that appear on the skin as small red dots or as linear or stellate lesions. They often progress to form papules, particularly on the face (Fig. 7).

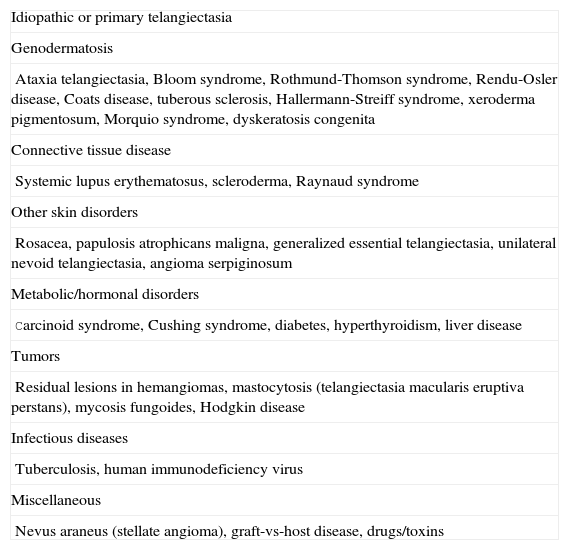
Although in theory, there are many possible causes for telangiectasia in childhood (Table 6), these lesions are uncommon. Most cases are idiopathic and take the form of small dot-like lesions that occur in association with congenital syndromes or collagen disorders. The lower eyelids are very frequently affected.
Causes of Telangiectasia in Childhood.
| Idiopathic or primary telangiectasia |
| Genodermatosis |
| Ataxia telangiectasia, Bloom syndrome, Rothmund-Thomson syndrome, Rendu-Osler disease, Coats disease, tuberous sclerosis, Hallermann-Streiff syndrome, xeroderma pigmentosum, Morquio syndrome, dyskeratosis congenita |
| Connective tissue disease |
| Systemic lupus erythematosus, scleroderma, Raynaud syndrome |
| Other skin disorders |
| Rosacea, papulosis atrophicans maligna, generalized essential telangiectasia, unilateral nevoid telangiectasia, angioma serpiginosum |
| Metabolic/hormonal disorders |
| Carcinoid syndrome, Cushing syndrome, diabetes, hyperthyroidism, liver disease |
| Tumors |
| Residual lesions in hemangiomas, mastocytosis (telangiectasia macularis eruptiva perstans), mycosis fungoides, Hodgkin disease |
| Infectious diseases |
| Tuberculosis, human immunodeficiency virus |
| Miscellaneous |
| Nevus araneus (stellate angioma), graft-vs-host disease, drugs/toxins |
Telangiectasia can be treated with all types of vascular-selective lasers, with excellent results.44 Certain forms of telangiectasia, however, are much more resistant to laser therapy. Examples include residual telangiectasia in hemangioma, unilateral nevoid telangiectasia, and telangiectasia associated with connective tissue disorders or Rendu-Osler-Weber syndrome.
Unilateral nevoid telangiectasia has a rare congenital form, which is more common in males, and an acquired form, which affects girls in puberty. The lesions tend to affect the face, neck, upper chest, and upper extremities.45
Ataxia telangiectasia, also known as Louis-Bar syndrome, is an autosomal recessive disorder caused by mutations in the ataxia telangiectasia mutated gene, ATM, at 11q22-23. This syndrome, which is characterized by telangiectasia (which tends to appear between the ages of 3 and 6 years), ataxia, and recurrent infections, is also associated with immune disorders, explaining why patients often develop reticuloendothelial neoplasms. A characteristic feature of this syndrome is bilateral conjunctival telangiectasia.46
Rendu-Osler-Weber syndrome, also known as hereditary hemorrhagic telangiectasia (HHT), is a hereditary condition characterized by cutaneous and mucosal telangiectasia and recurrent epistaxis. It is genetically heterogeneous and the mode of inheritance is autosomal dominant. The following genetic defects have been identified: mutations in ENG (9p33-34) in type 1 HHT, mutations in ACVRL1 (12q11-14) in type 2 HHT, a mutation at 5q31 in type 3 HHT, and a mutation at 7p14 in type 4 HHT. The skin lesions are uncommon in childhood and tend to appear after the second decade. They may be associated with arteriovenous malformations in the central nervous system, the bone marrow, the liver, the lungs, and the digestive system (15%-30% of cases).
AngiokeratomasAngiokeratomas are a heterogeneous group of lesions that present as red-violaceous to black papules with a wart-like surface (Fig. 8). Histologically, they are characterized by vascular dilatations in the papillary dermis with epidermal hyperplasia and marked hyperkeratosis. Their nomenclature has generated enormous debate in the literature.

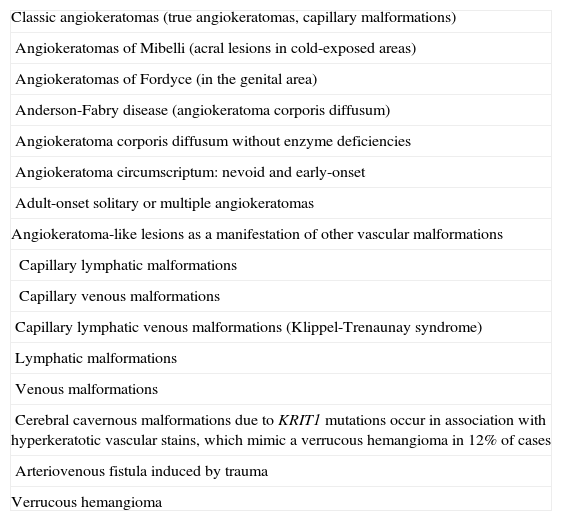
The main problem associated with the management of patients with angiokeratomas is that lesions that present clinically as angiokeratomas may have different underlying vascular malformations. Verrucous hemangioma, an as yet unclassifiable entity, is also a source of debate as it resembles an angiokeratoma but should probably be considered a separate entity. Table 7 shows a proposed classification of angiokeratoma-like lesions.
Classification of Angiokeratoma-Like Lesions.
| Classic angiokeratomas (true angiokeratomas, capillary malformations) |
| Angiokeratomas of Mibelli (acral lesions in cold-exposed areas) |
| Angiokeratomas of Fordyce (in the genital area) |
| Anderson-Fabry disease (angiokeratoma corporis diffusum) |
| Angiokeratoma corporis diffusum without enzyme deficiencies |
| Angiokeratoma circumscriptum: nevoid and early-onset |
| Adult-onset solitary or multiple angiokeratomas |
| Angiokeratoma-like lesions as a manifestation of other vascular malformations |
| Capillary lymphatic malformations |
| Capillary venous malformations |
| Capillary lymphatic venous malformations (Klippel-Trenaunay syndrome) |
| Lymphatic malformations |
| Venous malformations |
| Cerebral cavernous malformations due to KRIT1 mutations occur in association with hyperkeratotic vascular stains, which mimic a verrucous hemangioma in 12% of cases |
| Arteriovenous fistula induced by trauma |
| Verrucous hemangioma |
There are 5 classic types of angiokeratomas that can be considered true capillary malformations. These are angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma corporis diffusum, angiokeratoma circumscriptum, and acquired solitary or multiple angiokeratomas.
The treatment of choice is laser therapy. When pulsed-dye lasers are used, several sessions are generally required to eliminate the lesions completely due to the overlying hyperkeratosis. Fewer sessions are normally required with carbon dioxide (CO2) lasers or Nd:YAG lasers, currently the most widely used treatment modality.
Mibelli was the first person to describe an angiokeratoma when in the late 19th century he characterized the lesion that now carries his name. Angiokeratoma of Mibelli is associated with ischemia, and these lesions are therefore typically found on the fingers, toes, buttocks, knees, and ears. This form of angiokeratoma appears to be an autosomal dominant condition with variable penetrance. A subgroup of these lesions has been associated with connective disorders, and there have been some reports of angiokeratoma of Mibelli in childhood.47,48
Angiokeratoma of Fordyce affects the scrotum and the vulva. It has been linked to increased local venous pressure and is only rarely seen in childhood.
Angiokeratoma corporis diffusum was described by Anderson and Fabry at the end of the 19th century. This diffuse form of angiokeratoma was initially associated with α-galactosidase A deficiency, a metabolic disorder,49 but it is now believed to be possibly associated with 7 different enzyme deficiencies (Table 8). There have also been cases of angiokeratoma corporis diffusum in which no enzyme deficiencies have been detected, and cases of Fabry disease without angiokeratoma corporis diffusum and with just a few angiokeratomas or no skin lesions at all. There has also been a report of a patient with angiomas but without angiokeratomas.50 Most cases of Fabry disease are diagnosed on the basis of cardiac or renal manifestations in adulthood, when the disease is fully developed.51Unfortunately, only severe cases or cases in patients with a known family history of the disease are diagnosed in childhood. Angiokeratomas sometimes regress in patients with Fabry disease treated with enzyme replacement therapy.52
Enzyme Deficiencies Associated With Angiokeratoma Corporis Diffusum.
| α-Galactosidase A: Fabry disease |
| Sialidase: Sialidosis type II |
| β-Galactosidase and neuraminidase: galactosialidosis |
| α-Fucosidase: fucosidosis type II |
| α-N-acetylgalactosaminidase: aspartylglucosaminuria |
| or Kanzaki disease |
| β-mannosidase: mannosidosis |
| GM1 gangliosidosis |
Angiokeratoma circumscriptum, or angiokeratoma circumscriptum neviforme, is rare in childhood. It tends to present in patients in their mid or late 20s and most often affects the buttocks.53,54 It is more common in women and is considered to be a localized segmental form of Fabry disease.55,56
Finally, multiple or solitary acquired angiokeratoma lesions can occur at any site. This variant is also rare in childhood.
Verrucous hemangioma, which we have classified as a separate entity to angiokeratoma, is common in childhood.57 This rather inaptly named lesion has been the source of enormous controversy.58,59It is typically present at birth and is largely characterized by segmental lesions on the lower extremities (Fig. 9). It can, however, also present as isolated, medium-sized lesions at other sites. Verrucous hemangiomas are generally deep lesions that in some cases can even affect muscle. They often present very marked hyperkeratosis, which causes frequent episodes of bleeding. They share some of the features of malformations and vascular tumors. Histologically, they are interesting as over 50% of cases show GLUT1 positivity. Surgery is the treatment of choice for small lesions, but laser therapy should be considered for very large lesions, even if the goal is simply to alleviate symptoms.
Venous Malformations
Venous malformations are slow-flow vascular malformations composed of slow-growing, abnormally formed, dilated venous channels present at birth. They grow more quickly around puberty and following trauma.
They have been referred to by several names, including venous angioma, cavernous angiomas, and cavernous hemangiomas, but these terms should be avoided as they cause confusion.
For the sake of clarity, we have divided venous malformations into sporadic venous malformations, blue rubber bleb nevus syndrome, familial cutaneous and mucosal venous malformations, and glomuvenous malformations.
Sporadic Venous MalformationsSporadic venous malformations are bluish or violaceous masses that are easily compressed but regain their shape when released (Fig. 10). They also typically increase in size when venous pressure increases. They present as solitary or multiple lesions, generally have poorly circumscribed borders, and can be localized or diffuse, or superficial or deep. While they can affect any part of the body, they are most often found on the head and neck.

When pressed, there is no increased warmth or thrill, and the lesions may sometimes cause pain and swelling. These symptoms can appear spontaneously, generally due to the presence of thrombotic material in tortuous venous vessels, or they may be the result of trauma or hormonal changes. The presence of thrombotic material results in the formation of calcifications, known as phleboliths, that can be painful when pressed. The calcifications can be seen on a plain radiograph and are a very useful sign for confirming a diagnosis of venous malformations and ruling out other conditions.
When deep, the lesions can affect muscles, joints, and bones, and can even cause bone deformities or pathologic fractures. When they affect an entire extremity, they rarely cause severe progressive hemihypertrophy, unless they form part of a complex vascular malformation in Klippel-Trenaunay-Webner syndrome. Joint involvement can cause hemarthrosis and early arthrosis due to scarring processes. Muscle involvement can be a serious development because when nonextensible muscles are involved, such as those in the hand or foot, swelling caused by trauma can trigger a compartment syndrome.
Sporadic venous malformations present initially on the skin or mucous membranes. When the skin is affected, deeper involvement must be ruled out, as sometimes the visible mass is only the tip of the iceberg.
It is therefore essential to perform an imaging study, generally magnetic resonance angiography, to determine the extent of the lesion with the greatest precision possible.
Spontaneous venous malformations exhibit slow, progressive growth of both superficial and deep structures. Rapid growth, however, can be triggered by intralesional bleeding.
Patients with venous malformations and extensive muscle involvement often have chronic coagulopathy, a condition that was, for decades, misdiagnosed as thrombocytopenia related to Kassabach-Merritt syndrome.60 It is important to recognize this chronic localized intravascular coagulopathy, which can cause acute pain, as it can lead to serious problems in patients undergoing surgery for other reasons.61,62 Localized intravascular coagulopathy is characterized by elevated D-dimer levels (the main marker used to monitor the course of disease) and normal fibrinogen levels.63 Coagulation disorders are rare in childhood and tend to occur in adults with untreated malformations.
It was recently reported that, over time, patients with venous malformations and coagulation disorders can develop pulmonary hypertension due to the repeated release of small clots that lodge in the lungs without initially causing symptoms.64
In 2008, Zietz et al.65 described a variant of sporadic venous malformations that they called venous nevus or nevus venosus. These sporadic solitary lesions are arranged in a segmental pattern attributable to genetic mosaicism.
Sporadic venous malformations are treated with pressure garments, surgery, laser therapy, and sclerotherapy. The treatment of choice is surgery, but this is only performed with curative intent in isolated cases as venous malformations tend to be invasive and surgery would be highly mutilating.66 Where possible, compression therapy should be initiated to delay lesion growth and relieve symptoms. Percutaneous image-guided sclerotherapy can be performed with alcohols such as ethanol, bleomycin, and ethoxysclerol; excellent results have also been recently reported for polidocanol foam.67,68 Repeated sessions under general anesthesia are necessary and occasionally the size of the lesion is reduced enough to allow surgery. Laser therapy should be reserved for superficial lesions (Nd:YAG lasers)69 or palliative treatment of the surface of a deeper malformation (CO2 or Nd:YAG lasers). There have also been recent reports of endovenous diode laser therapy used either alone or in combination with other treatments such as sclerotherapy.70
Blue Rubber Bleb Nevus SyndromeBlue rubber bleb nevus syndrome, also known as Bean syndrome, consists of typical venous lesions with 2 main characteristics: 1) they present as multiple lesions, although large solitary masses, frequently located at the cephalic pole, are occasionally seen at birth; and 2) they occur in association with visceral lesions that affect the gastrointestinal system in particular. Patients thus often have gross or occult blood in the stools and iron deficiency anemia71 (Fig. 11). There may also be neurologic symptoms,72sometimes as a primary manifestation. The syndrome has also been associated with multiple intracranial vascular malformations, developmental venous anomalies in the brain, and sinus pericranii.73
, which is characterized by the association of multiple visceral lesions, mostly affecting the digestive system.")
Blue rubber bleb nevus syndrome is a rare condition of unknown etiology that normally presents sporadically, although it can be inherited as an autosomal dominant trait.
The lesions increase in number and size with age, and in severe cases gastrointestinal hemorrhage can lead to death.74 A recent report described how intestinal blue rubber bleb nevus lesions were studied by capsule endoscopy.75
The lesions are normally always present in childhood, but tend only to cause significant symptoms in very severe cases.76
As occurs in sporadic venous malformations, patients may also have coagulopathy77 with severe hypofibrinogenemia.
Distinguishing rubber bleb nevus syndrome lesions from other venous lesions such as glomuvenous lesions78 can be difficult. Dermoscopy can be useful79 but the diagnosis must be confirmed histologically.
Visceral lesions are treated surgically via enteroscopy and argon plasma coagulation, and there have been reports of cases in which dozens of intestinal resections eradicated these lesions and prevented recurrence.80,81 There have also been descriptions of lesions being treated with etidronate82 or polidocanol foam sclerotherapy,83 although such treatment was only effective in isolated lesions.
Familial Cutaneous and Mucosal Venous MalformationsFamilial cutaneous and mucosal venous malformations are caused by a mutation in the TEK gene at chromosome 9p21-22, and the pattern of inheritance is autosomal dominant.84–86 TEK is an endothelial cell tyrosine kinase receptor that binds angiopietin-1. It plays a very important role in vasculogenesis and angiogenesis, as shown by the fact that mice deficient for TEK develop serious vascular malformations.87 When altered by a mutation, the TEK receptor induces ligand-independent activation, presumably suppressing apoptosis of endothelial cells.
The lesions are clinically indistinguishable from sporadic venous malformations, and therefore all the considerations described above for that type of anomaly also apply to these familial malformations. Indeed, sporadic venous malformations have also been found to have TEK mutations.86
Glomuvenous MalformationsGlomuvenous malformations are uncommon malformations that originate in the glomus cells.88 These lesions used to be called glomangiomas, but they are actually malformations and not tumor lesions.
They can occur sporadically, but they are mostly inherited in a paradominant pattern; the altered protein is glomulin, which is encoded by a gene localized to chromosome 1p 21-22. Unlike the venous malformations described above, glomuvenous malformations tend to be painful when pressed and only affect the skin and subcutaneous cell tissue; they do not affect mucous membranes, bones, muscles, or joints. They can usually be clinically distinguished from other venous malformations as they are firmer, more solid, and not readily compressed.
There are 3 clinical variants: 1) a solitary form that accounts for 90% of all lesions and is typically located on the nail bed; 2) a multiple form that is familial in most cases and can manifest as extensive or segmental lesions; and 3) a congenital plaque variant with atypical presentation.89
The mainstay treatment for glomuvenous malformations is surgery, but long-pulsed Nd:YAG lasers that penetrate only a few millimeters below the skin are a very interesting option for small, isolated lesions.
Finally, an association between cutaneous and mucosal venous malformations and familial cerebral cavernous malformations related to the KRIT1 gene has been described.90 These patients may also have capillary malformations or angiokeratoma-like lesions on the skin.
Lymphatic MalformationsThe lymphatic system plays an essential role in fat absorption, the immune response and in maintaining fluid balance. Abnormalities in the embryonic development of the lymphatic system give rise to lymphatic malformations.91
The terminology used to refer to these slow-flow vascular malformations has generated enormous confusion.92
There are 2 categories of lymphatic malformations that affect the skin: lymphedema and congenital lymphatic malformations. Lymphedema can be primary or secondary. Primary lymphedema is caused by hypoplasia of the lymph trunks or lymph nodes while secondary lymphedema is caused by damage to the lymphatic system. Congenital lymphatic malformations can be superficial or deep and present as solitary or multiple cystic malformations. Cystic lesions are described as macrocystic (formerly called cystic hygromas), microcystic, or mixed.93 Microcystic lymphatic malformations have been referred to as lymphangioma, lymphangioma circumscriptum, lymphangioma simplex, verrucous hemangioma, and angiokeratoma circumscriptum.94 Finally, lymphatic malformations can occur in a large number of syndromes (Table 9).
Syndromes Associated With Lymphatic Malformations.
| Gorham disease |
| Hennekam syndrome |
| Intestinal lymphangiectasis |
| Klinefelter syndrome |
| Klippel-Trenaunay-Webner syndrome |
| Lymphedema-distichiasis |
| Lymphedema-hypoparathyroidism |
| Meige disease (lymphedema tarda) |
| Milroy disease |
| Neurofibromatosis |
| Protein-losing enteropathy |
| Stewart-Treves syndrome |
| Triploid syndrome |
| Turner syndrome |
| Yellow nail syndrome |
| Noonan syndrome |
| Edwards syndrome |
| Maffucci disease |
| Patau syndrome |
| Aagenaes syndrome |
| Prader-Willi syndrome |
| Lymphangioleiomyomatosis |
| Pulmonary lymphangiectasia |
| Blue rubber bleb nevus syndrome |
| Proteus syndrome |
| Adams-Oliver syndrome |
| Microcephaly-lymphedema-chorioretinal dysplasia |
| Emberger syndrome |
| CLOVESa syndrome |
| CLAPOb syndrome |
No genetic associations have been demonstrated for lymphatic malformations. The lesions occur sporadically, and if there are genetic causes, they are probably somatic mutations as germinal cell mutations would be fatal.95
The term lymphangiectasia is used to refer to pseudovesicular lesions filled with a clear fluid, although some blood may be present if the lesions are connected to the venous system. It may be related to lymphatic malformations or alterations caused by these malformations.
These lesions are always present as birth, but may not become evident until years later. They grow in proportion to the child's somatic growth and frequently become inflamed due to infections or trauma. Such inflammation, which is occasionally severe, can cause serious problems because the lesions are frequently located on the head and neck.
Lymph nodes form during both embryonic development and in adulthood. Lymphangiogenesis is poorly understood, but the role of vascular endothelial growth factor (VEGF) C96 and TIE197 appears to be important. In fact, mice deficient for TIE1 develop serious lymphatic system alterations. Numerous lymph vessel markers have emerged in recent years. Of these, the following are of particular interest: lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1)98 and prospero homeobox protein 1 (PROX1),99 which are both expressed by embryonic lymphatic vessels; VEGF receptor 3 (VEGFR-3), which is initially present in all embryonic vessels but later only in lymphatic vessels and is essential for the growth and maintenance of lymphatic endothelial cells100; and VEGF C and VEGF-D, which are the main ligands of VEGFR-3 and VEGFR-2.
Macrocystic Lymphatic MalformationsThese large cystic lesions, which increase in size over time, form when the major lymph sacs fail to connect to the venous system during embryonic development; the lesions were formerly known as cystic hygromas.101 If the lymphatic and venous structures finally connect, the cysts disappear, but otherwise they form macrocystic lymphatic malformations, which often occur in association with microcystic lesions.
They normally occur on the neck, in the axillas, or on the lateral aspect of the trunk and they can be solitary or multiple, and may or may not be interconnected. The overlying skin generally has a normal appearance and the cysts can reach a considerable size as they grow. They are present at birth in 60% of cases and are diagnosed before 2 years of age in 85% of cases.
The most serious complication is bleeding, which can result in extensive acute inflammation that can block the airways or cause serious eye disorders, depending on the location of the lesions. On occasions, inflammation is the first sign of a malformation.102
Macrocystic lymphatic malformations are generally easy to detect with a simple ultrasound scan, but pleural effusion and ascites must always be ruled out.
The initial treatment of choice is percutaneous sclerotherapy, and in children excellent results have been achieved with OK-472,103 doxycycline, and bleomycin. When sclerotherapy fails, surgery can resolve or alleviate the condition. Improvements in both symptoms and complications are proportional to the reduction achieved in the size of the lymphatic mass.104
Microcystic Lymphatic MalformationsMicrocystic lymphatic malformations develop when the distal lymph vessels fail to connect to the lymph channels during embryonic development. This disorder is still commonly referred to as lymphangioma circumscriptum in the literature. Because these structures do not form part of the general lymphatic system, microcystic lesions do not generally give rise to lymphedema.
Their clinical presentation is much more varied than that of macrocystic lesions. The most common presentation is lymphangiectasias in the form of small translucent pseudovesicles containing a clear fluid; this can occur in any location, including the mucous membranes (Fig. 12). Sometimes, when the malformation is connected to the venous system (with which it has a common embryologic origin) the vesicles fill with blood and acquire a red-purple color. Clusters of vesicles often form plaques over the underlying malformation, but the size of the superficial lesion is not a good indicator of the depth of the underlying malformation. This can only be determined by imaging studies. It is also important to note that these vesicles can also appear on the surface of secondary lymphatic lesions, such as lymphedema.

In our experience, the co-occurrence of a capillary malformation is a sign of poor prognosis, aggressive behavior, and invasion.
The cysts can also take the form of verrucous lesions, simulating warts or angiokeratoma-like lesions against a violaceous background, explaining why this malformation has sometimes been described as angiokeratoma circumscriptum. When the lesions are located deep in the papillary dermis or in the subcutaneous tissue, the only visible sign may be a small purple mark on the epidermis during inflammatory episodes. They have, thus, on occasions, been confused with bruises from suspected child abuse.
Ultrasound can be extremely useful for diagnosis, but magnetic resonance imaging is probably of more value as it can determine the extent of the lesions.105,106
Microcystic lymphatic malformations are treated surgically. Surgery tends to be palliative because the lesions are highly invasive and complete excision without sequelae is generally difficult as small vesicles are rarely connected and therefore do not permit the distribution of the sclerosing agent. Localized lesions in the oral cavity or on the tongue can be treated using palliative CO2 laser therapy107 or long-pulsed Nd:YAG laser therapy. Pulsed-dye lasers have also been used,108 and a recent report describes the treatment of microcystic lymphatic malformations with a combination of radiofrequency ablation and sclerotherapy.109
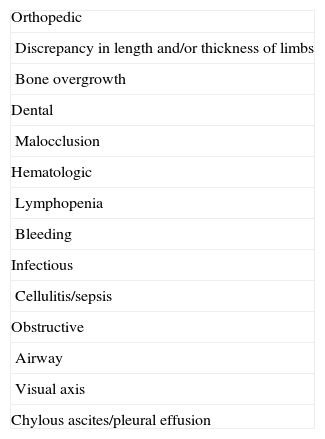
The complications associated with these lesions are summarized in Table 10.
LymphedemaLymphedema is the most common lymphatic disorder in adults, but its presence in children should suggest a primary lymphedema (one caused by a developmental defect affecting the lymphatic system) (Fig. 13).110 Milroy disease is a congenital lymphedema characterized by the presence of unilateral or bilateral swelling of the legs, the arms, and/or the face, with progressive, irreversible fibrotic changes (mutation in the 5q34-35 region affecting the VEGFR-3 gene111). Meige disease is a lymphedema that presents in puberty, often in association with episodes of cellulitis.112 Another type of lymphedema that appears in puberty has been reported in association with distichiasis (double rows of eyelashes); it is caused by haploinsufficiency in FOXC2, a gene that encodes the nuclear transcription factor.113 Hypotrichosis-lymphedema-telangiectasia has been linked to the SOX18 gene.114
.")
Other forms of childhood lymphedema form part of various syndromes, including Turner syndrome, Noonan syndrome, and Klinefelter syndrome, all of which will be discussed in the second part of this review.
Secondary lymphedema in childhood is normally a result of severe skin infections that lead to lymphadenitis, abscesses, and induration of the inguinal lymphatics, with functional loss.
Future TreatmentsThe exponential progress made in molecular biology in recent years has created high expectations regarding the future treatment of lymphatic lesions. In animal models, it has been shown that treatment with recombinant VEGF-C induces growth of the lymphatic capillary network and improves the symptoms of secondary lymphedema.115,116 It has also been shown that lymphoangiogenesis induced by recombinant factors induces the regeneration of lymph vessel valves. Finally, the transplant of lymph nodes with VEGF-C has been found to restore the anatomy of the lymphatic network, including lymph vessels and nodes, in defective areas.117
Fast-Flow Vascular MalformationsFast-flow vascular malformations occur in arterial lesions or arteriovenous lesions, which form when veins are connected to arteries without an intervening capillary bed. In other words, the blood flows directly from a high-pressure system to a low-pressure system. This causes very fast blood flow that can be detected by Doppler ultrasound. When the flow is very intense, it can be felt clinically as a thrill or pulsation.
Fast-flow malformations include arterial malformations, arteriovenous fistulas, and arteriovenous malformations. Arteriovenous malformations can occur alone or form part of a wide range of syndromes (Table 11).
Arteriovenous Malformations: Associated Syndromes.
| Bonnet-Dechaume-Blanc syndrome (arteriovenous malformations of the retina and choroid) |
| Wyburn-Mason syndrome (neurologic and mental abnormalities |
| Brégeat syndrome (arteriovenous malformations of the conjunctiva, cheeks, nose, and mouth) |
| Cobb syndrome (medullary arteriovenous malformations occurring at the same metamere) |
| Parkes-Weber syndrome: arteriovenous malformations in a limb |
| Rendu-Osler disease |
| Hereditary hemorrhagic telangiectasia |
| Cowden disease |
| Neurofibromatosis |
| Epithelioid hemangioma |
| Bluefarb-Stewart syndrome |
| Diffuse dermal angiomatosis |
| Lhermitte-Duclos syndrome |
| Acroangiodermatitis of Mali |
| Angiolymphoid hyperplasia with eosinophilia |
| Reactive angioendotheliomatosis |
| Angiokeratoma corporis diffusum without Fabry disease |
| Capillary malformations: RASA1 mutations |
| Multiple cutaneous follicular hamartomas |
| Multiple glomus tumors |
| Facial hemangiomas |
| Aplasia cutis congenita |
| Linear sebaceous nevus syndrome |
Arterial malformations, such as aneurysms, stenosis, aplasia, and ectasia rarely occur in the skin. Direct arteriovenous fistulas, unlike central nervous system arteriovenous malformations, are rarely cutaneous and are almost always the result of trauma. Arteriovenous malformations, which can be localized or extensive, are therefore the only fast-flow vascular malformations that are found in the skin.
Compared with slow-flow malformations, arteriovenous malformations are rare in childhood. The most common arteriovenous malformations are cerebral malformations, which are not addressed in this review. While cutaneous arteriovenous malformations are always present at birth, they rarely exhibit congenital symptoms or symptoms during the first years of life. They are a major diagnostic challenge in childhood and are generally only diagnosed correctly when fully developed.
Schobinger devised a clinical staging system for arteriovenous malformations that described 4 stages of development. Stage 1 lesions tend to be pink macules or patches that mimic a simple capillary malformation (Fig. 14), but thrill and increased warmth suggest the presence of a fast-flow component. They are generally asymptomatic and remain so until puberty, although some patients never develop symptoms. Stage II lesions are more prominent and have greater vascular dilation. They are characterized by increased thrill and pulsatility and have more dilated drainage veins; in some cases, the overlying skin acquires a violaceous tone. Stage I lesions tend to progress to stage II lesions in puberty as a result of hormonal changes, trauma, or partial treatment. Stage III is characterized by destruction of the deep tissues, including the bone, and the presence of pain and bleeding. Finally, stage IV is characterized by an increase in blood flow that induces cardiac decompensation.

Arteriovenous malformations have a broad clinical spectrum (Table 12) and cause a wide range of symptoms, including functional limitations, pain (in over 50% of cases), hyperhidrosis, hypertrichosis, hyperemia, thrill, trophic changes, ulceration, bleeding, and pseudo-Kaposi lesions.118 They are generally the most difficult type of vascular malformations to diagnose.
Arteriovenous Malformations: Clinical Manifestations.
| Congenital salmon patch |
| Dilated veins, often with thrill |
| Palpable thrill |
| Pulsatile lesions |
| One extremity that gradually becomes longer than the other until the end of puberty |
| Diffuse hypertrophy with an increase in the length of the affected limb |
| Localized soft tissue hypertrophy |
| Lymphedema and lymphangiectasias |
| Pigmentation |
| Foot swelling |
| Fibrosis |
| Ulcers |
| Pseudo-Kaposi sarcoma lesions (Stewart-Bluefarb syndrome) |
| Lytic bone lesions |
| Heart failure with multiple arteriovenous fistulas |
Doppler ultrasound is important for diagnosis, as is MRI, which is useful for determining the extent of the lesion. Imaging studies, preferably computed tomography angiography, must be performed before treatment is initiated to determine the morphology and size of the tangle of abnormal arteries and veins we call the nidus.
The management of arteriovenous malformations is also a major challenge and requires a multidisciplinary approach, although in many cases, this does not guarantee adequate management. Arteriovenous malformations exhibit unpredictable behavior and are associated with a high rate of recurrence, regardless of the treatment modality.119 Embolization should be performed 24 to 48hours before surgery to achieve the maximum reduction possible in the size of the malformation. The goal of surgery is to eradicate all or as much of the lesion as possible, while minimizing morbidity. Success hinges on the elimination of the nidus. Because arteriovenous malformations tend to contain large vessels, presurgical embolization is important as it significantly reduces intraoperative bleeding.
Since arteriovenous malformations with skin involvement classified as Schobinger stage II or higher are rarely seen in childhood, treatment at this age is rare.
Complex-Combined Vascular MalformationsAs mentioned earlier, the ISSVA vascular malformation classification separates lesions according to the predominant type of vessel affected, but in many cases, there may be several types of predominant vessels. These are known as complex-combined vascular malformations.
Capillary lymphatic malformations tend to present as a port-wine stain containing angiokeratoma-like lesions that correspond to the lymphatic component (Fig. 15). Some deep lymphatic malformations in the abdominal cavity also have an associated superficial capillary malformation. When a capillary lymphatic malformation is suspected, it is important to perform imaging studies to search for deep lesions.

Capillary venous malformations also present as port-wine stains, but they contain prominent veins rather than angiokeratoma-like lesions. They often occur on the lower extremities without hypertrophy of the affected extremity.
As the prognosis is different, it is important to differentiate them from lesions seen in Klippel-Trenaunay-Webner syndrome, a condition characterized by a capillary malformation, venous malformations, and lymphatic malformations with hemihypertrophy. There have also been a small number of reports of limited arteriovenous malformations in Klippel-Trenaunay-Webner syndrome.
The combination of lymphatic and venous malformations generally has a worse prognosis than either of these malformations in isolation. Where possible, surgery is the treatment of choice.
Capillary lymphatic venous malformations are almost always seen in Klippel-Trenaunay-Webner syndrome. The presence of associated lymphatic malformations is a sign of worsening disease and poor prognosis.
Parkes-Weber syndrome is characterized by capillary malformations, arteriovenous malformations, and progressive hemihypertrophy (Fig. 16). The syndrome is rare and has a poor prognosis. Multiple cutaneous capillary malformations—generally hereditary—also occur in association with cerebral arteriovenous malformations. Both types of malformations have been linked to RASA1, a gene that has also been linked to many cases of Parkes-Weber syndrome. Mutations in this gene have also been linked to cases of familial capillary malformations without arteriovenous malformations.
.")
The combined presence of capillary, venous, arteriovenous, and lymphatic malformations has also been recently described in CLOVES syndrome, which involves congenital (C), lipomatous overgrowth (LO), vascular malformations (V), epidermal nevi (E), and scoliosis/skeletal/spinal anomalies (S).
Vascular Malformations Associated With Vascular TumorsVascular malformations have also been reported in association with vascular tumors in childhood. Examples are congenital hemangioma and capillary malformation,120 infantile hemangioma and cutaneous marmorata telangiectatica congenita, pyogenic granuloma and capillary malformations121 or arteriovenous malformations,122 segmental hemangiomas and arterial anomalies (PHACE syndrome),123 hemangioendothelioma with spindle cells and venous malformations,124 and kaposiform hemangioendothelioma and lymphatic malformation.125
To conclude, vascular malformations are complex and often rare conditions that should be managed by multidisciplinary teams at vascular anomalies centers, as is currently being done in several speciality hospitals.
Conflict of InterestsThe authors declare that they have no conflicts of interest.
Please cite this article as: Del Pozo J, Gómez-Tellado M, López-Gutiérrez JC. Malformaciones vasculares en la infancia. Actas Dermosifiliogr.2012;103:661-678.



