





Epidermolysis bullosa acquisita (EBA) is an autoimmune subepidermal blistering disease caused by autoantibodies to type VII collagen. The clinical presentation is variable, with skin and mucosal lesions that can cause significant dysfunction. Different treatment options exist, but the results are often unsatisfactory.
ObjectiveTo review all the cases of epidermolysis bullosa acquisita (EBA) diagnosed at our hospital over a 26-year period.
Materials and methodsWe performed a retrospective review of the clinical, histologic, and immunologic features of EBA in 9 patients.
ResultsMean age at presentation was 37 years and 66.67% of the patients were women. EBA occurred in association with malignant tumors, inflammatory bowel disease, and autoimmune disorders. The most common variant was inflammatory EBA (6 of the 9 cases). In all 9 patients, histology revealed a subepidermal blister and direct immunofluorescence showed linear deposits of immunoglobulin G and C3 in the basement membrane zone. Indirect immunofluorescence performed on salt-split skin substrate was positive in 6 patients and showed a dermal pattern in all cases. Five patients were tested for autoantibodies to type VII collagen using enzyme-linked immunosorbent assay, with positive results in 2 cases. Immunoblotting using recombinant noncollagenous domains (NC1) of type VII collagen was positive in all 6 cases in which it was performed. Response to treatment was variable.
ConclusionsEBA is a rare disease with a variable clinical presentation that can be confused with that of other subepidermal blistering diseases. Correct diagnosis requires a high level of clinical suspicion and the use of all available diagnostic tests. Thorough evaluation of cutaneous and mucosal involvement and prompt initiation of appropriate treatment will ensure the detection and prevention of dysfunction and treatment-related complications.
La epidermolisis ampollosa adquirida es una enfermedad ampollosa subepidérmica autoinmune causada por autoanticuerpos contra el colágeno VII. Su clínica es heterogénea con afectación de piel y mucosas pudiendo generar secuelas invalidantes. Existen diversas opciones terapéuticas frecuentemente insatisfactorias.
ObjetivoRevisar los casos de epidermolisis ampollosa adquirida diagnosticados durante un periodo de 26 años.
Material y métodosEstudio retrospectivo de las características clínicas e inmunopatológicas de 9 pacientes con dicho diagnóstico
ResultadosLa mediana de edad de presentación fue de 37 años, el 66.67% de pacientes fueron mujeres. Asociaciones: neoplasias malignas, enfermedad inflamatoria intestinal y procesos autoinmunes. La variante inflamatoria fue la más frecuente (6/9). La histología mostró constantemente una ampolla subepidérmica y la inmunofluorescencia directa la presencia de depósitos lineales de IgG y C3 en membrana basal. La inmunofluorescencia indirecta fue positiva en 6 pacientes, mostrando en todos ellos un patrón dérmico en piel separada. En 5 pacientes se determinaron los anticuerpos contra el colágeno VII por Enzyme-Linked Immuno Sorbent Assay de los cuales 2 fueron positivos, e Inmunoblot con NC1 recombinante en 6 casos, positivo en todos ellos. La respuesta terapéutica fue variable.
ConclusionesSe trata de una enfermedad rara, de clínica heterogénea que puede inducir a confusión con otras enfermedades ampollosas subepidérmicas. Se requiere un alto índice de sospecha y el empleo de todos los métodos disponibles para establecer su diagnóstico. La correcta evaluación de la afectación cutáneo-mucosa y la instauración precoz de la terapéutica adecuada permitirá la detección de sus secuelas y de las complicaciones del tratamiento.
Epidermolysis bullosa acquisita (EBA) is a chronic subepidermal bullous disease of the cutaneous and mucosal tissues.1–3 EBA was originally described by Roenigk and colleagues1 in 1971 as a mechanobullous disorder associated with skin fragility. Although similar to congenital epidermolysis bullosa, EBA appears in adults with no family history. In 1973 Kushniruk4 reported that direct immunofluorescence (DIF) revealed immunoglobulin (Ig) G and C3 deposition in the basement membrane of tissue samples from patients with EBA. In 1981 Yaoita and colleagues5 used immunoelectron microscopy techniques to demonstrate the location of deposits in the sublamina densa in EBA and in the lamina lucida in bullous pemphigoid. In 1984 Woodley and colleagues6 identified a 290-kDa protein as the target antigen in EBA; this protein was later identified as type VII collagen.7 An inflammatory variant of this disorder was then described; clinical characteristics were similar to those of bullous pemphigoid and other pemphigoid subtypes, mimicking mucosal pemphigoid or Brunsting-Perry cicatricial pemphigoid.8–10 Features of both conditions may be present at once or the clinical picture may shift toward one or the other during the course of disease.11
EBA is an uncommon disease with an estimated incidence of 0.2 cases per million inhabitants.2,3 The low prevalence has meant that no racial predilection has been established, although black patients of African descent were reported to be overrepresented in one patient series12 and higher prevalence has also been described for Korean populations.13 Age at onset is usually between 40 and 50 years, although EBA has been found in patients of advanced age and in children.14,15 Vertical transmission from a mother with EBA to an infant has also been reported.16
The etiology is unknown but it seems clear that autoantibodies to type VII collagen play a role in the pathogenesis of this disease. The relevance of these antibodies has been demonstrated by the development of blisters in an infant as a result of the transfer of antibodies from maternal blood to the fetus.16 Ex vivo evidence comes from the induction of dermal-epidermal separation, with neutrophil recruitment, in frozen sections of human skin exposed to sera from patients with EBA.17 Experimental in vivo models of EBA have been based on passive immunization (transfer to mice of IgG antibodies to type VII human or rabbit collagen)18 or active immunization (of certain mice strains with immunodominant fragments in the NC1 domain of type VII collagen).19
The classic mechanobullous form of EBA is characterized histologically by the presence of subepidermal blisters with scant inflammatory infiltrate. Inflammatory EBA is marked by a more intense infiltrate containing neutrophils in abundance and occasionally eosinophils. DIF reveals linear, predominantly IgG deposition along the dermal-epidermal basement membrane. C3, IgA, or IgM deposition of variable intensity is observed in some cases.4,5,20 Indirect immunofluorescence (IIF) of sera from some patients may detect the presence of circulating IgG autoantibodies that target the dermal-epidermal basement membrane. It is possible to use different substrates, such as human skin or monkey esophageal tissue,5 but the best results are obtained using human skin separated in a 1.0-M sodium chloride (NaCl) solution. The use of salt-split skin enables detection of circulating IgG autoantibodies that bind to the dermal (base) side of the separation, a finding that distinguishes EBA from bullous pemphigoid, in which these autoantibodies bind to the epidermal (roof) side. Although this is a relatively simple way to differentiate these 2 diseases, it will not distinguish EBA from other autoimmune subepidermal bullous diseases with a dermal pattern, such as bullous systemic lupus erythematosus (BLSE) or various forms of pemphigoid associated with anti-laminin 332, anti-laminin γ1 or anti-laminin-p 105).21 When IIF is negative, DIF can be used after inducing separations in biopsy material (with the 1.0-M NaCl solution); alternatively, a suction-induced blister can be biopsied for DIF. DIF will also pinpoint the location of IgG deposits in the dermal portion of the separation in EBA. However, like IIF, this technique will not differentiate between EBA and other diseases with a dermal pattern.22 The diagnostic value of the pattern of autoantibody deposition in the membrane has recently been described. When IgG deposits form above the sublamina densa (in bullous pemphigoid and anti-lamina 332 pemphigoid), an n-serrated pattern is typical; when deposits are in the sublamina densa (as in EBA), a u-serrated pattern forms; for this technique sections must be of less than 4μm.23
Immunoblotting of serum from patients with EBA using dermal extracts identifies protein bands of approximately 290 kDa and 145 kDa, corresponding to complete (dimeric) type VII collagen or one of its regions (monomeric). Most antigen epitopes of collagen VII are located in the NC1 domain (145 kDa). Therefore, recombinant NC1 protein produced in the laboratory can also be used in immunoblot analysis or in classic enzyme-linked immunosorbent assay (ELISA). ELISA using recombinant NC1 is considerably more sensitive and specific than IIF on a split-skin substrate or immunoblotting with dermal extracts; ELISA also quantifies autoantibodies.24 A recently desribed technique using complete type VII collagen (containing both the NC1 and NC2 domains) has been shown to be slightly more sensitive than the technique using only NC1.25
Immunoelectron microscopy is the gold standard diagnostic method because it identifies the location of immunoglobulin deposition in the sublamina densa in EBA and in the lamina lucida and hemidesmosomes in bullous pemphigoid.5,20 However, this is a complex technique that is unavailable in many centers.
EBA can be diagnosed with a fair degree of certainty by correlating clinical findings with the results of the various diagnostic tools. However, it cannot be distinguished from BSLE in this way, since pathogenic autoantibodies to type VII collagen are present in both diseases.26 For a diagnosis of BSLE, the American College of Rheumatology (ACR) criteria must be met. BSLE lesions are mainly located in skin areas exposed to sunlight, and this disease is less refractory to treatment than EBA.27
Various therapies for EBA have been tried and have often proven ineffective; in any case, response is unpredictable. Few large patient series have been published and no randomized clinical trials have been performed because the disease is so rare.3,28
We analyzed the demographic, clinical, and immunopathologic characteristics of EBA as well as response to therapy in a series of patients diagnosed between 1985 and 2011 in our hospital.
Patients and MethodsThis retrospective study included patients diagnosed with EBA in Hospital Clínic de Barcelona between 1985 and 2011. Diagnosis was based on clinical, histologic, and immunopathologic evidence. We analyzed all demographic, clinical, immunopathologic, and therapeutic data on record.
For IIF studies we used salt-split human skin prepared conventionally by incubation in a 1.0M NaCl solution; patient serum samples for studies were obtained during the active phase of disease.21 For DIF we used the method described by Gammon and colleagues.22 Immunoblotting was performed using dermal extracts and, in some cases, recombinant proteins of the NC1 domain of type VII collagen were used. ELISA was performed on domain-specific recombinant proteins (His-hCVII-NC1 and NC2-His-hCVII-NC1 NC2-H) of type VII collagen, as described elsewhere.25
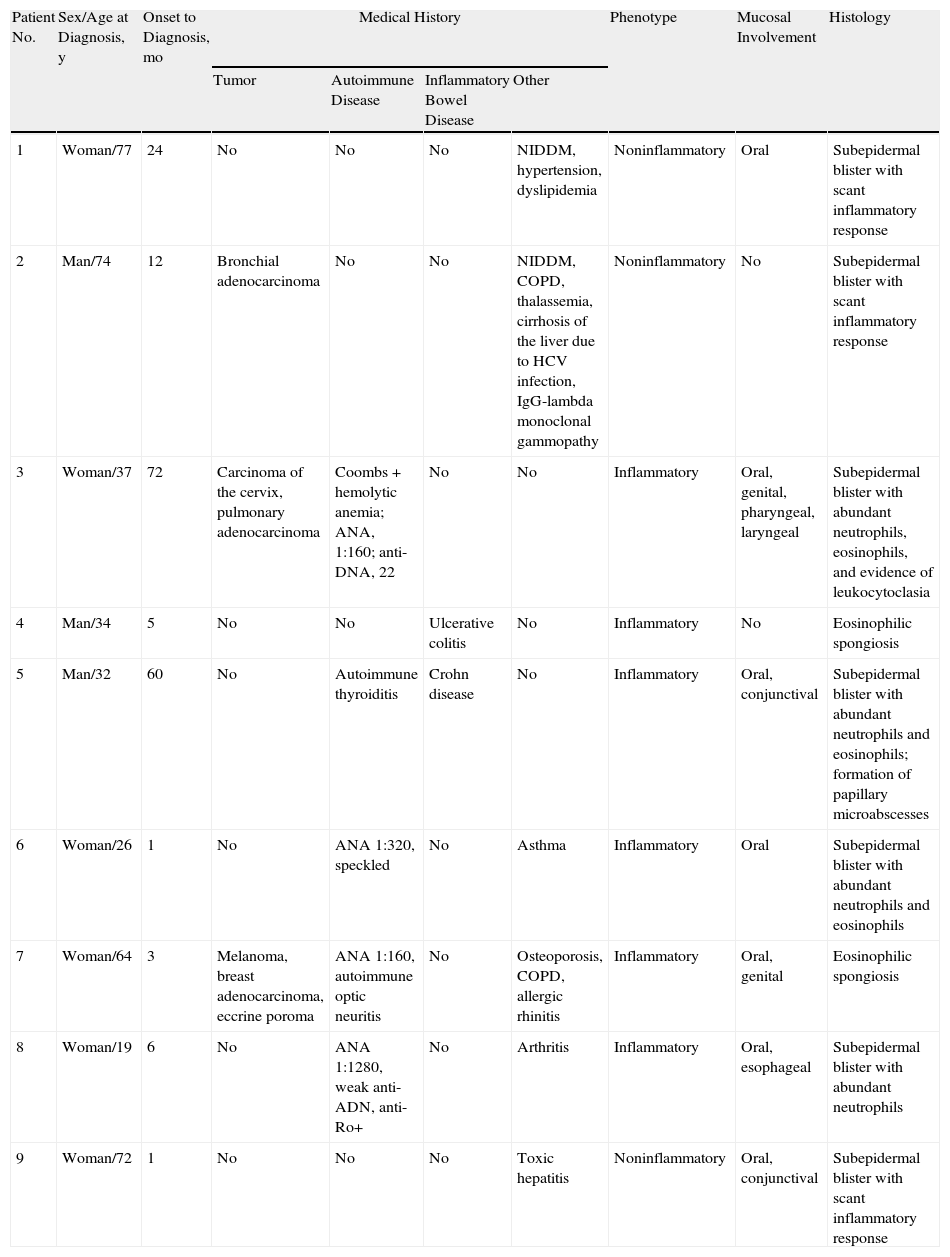
ResultsWe identified 9 patients diagnosed with EBA between 1985 and 2011. The mean age was 37 years; 3 were men and 6 women (male:female ratio, 1:2). The median time between onset and diagnosis was 7 months (range, 1–72 months).
Most of the patients (6 out of 9) presented with the inflammatory form (Fig. 1). The classic mechanobullous form (Fig. 2) was seen in patients of more advanced age: all patients with this form were over 72 years old. In 1 patient (Case 5, Table 1), the clinical phenotype changed from bullous pemphigoid to Brunsting-Perry pemphigoid. Disease was confined to the skin in only 2 patients. All of the 7 patients with mucosal involvement had oral lesions; additionally, 2 had lesions on the genitals, 2 had conjunctival involvement, and 1 had lesions on the pharynx, larynx and esophagus.


Epidermolysis Bullosa Acquisita: Clinical and Immunopathologic Characteristics.
| Patient No. | Sex/Age at Diagnosis, y | Onset to Diagnosis, mo | Medical History | Phenotype | Mucosal Involvement | Histology | |||
| Tumor | Autoimmune Disease | Inflammatory Bowel Disease | Other | ||||||
| 1 | Woman/77 | 24 | No | No | No | NIDDM, hypertension, dyslipidemia | Noninflammatory | Oral | Subepidermal blister with scant inflammatory response |
| 2 | Man/74 | 12 | Bronchial adenocarcinoma | No | No | NIDDM, COPD, thalassemia, cirrhosis of the liver due to HCV infection, IgG-lambda monoclonal gammopathy | Noninflammatory | No | Subepidermal blister with scant inflammatory response |
| 3 | Woman/37 | 72 | Carcinoma of the cervix, pulmonary adenocarcinoma | Coombs+hemolytic anemia; ANA, 1:160; anti-DNA, 22 | No | No | Inflammatory | Oral, genital, pharyngeal, laryngeal | Subepidermal blister with abundant neutrophils, eosinophils, and evidence of leukocytoclasia |
| 4 | Man/34 | 5 | No | No | Ulcerative colitis | No | Inflammatory | No | Eosinophilic spongiosis |
| 5 | Man/32 | 60 | No | Autoimmune thyroiditis | Crohn disease | No | Inflammatory | Oral, conjunctival | Subepidermal blister with abundant neutrophils and eosinophils; formation of papillary microabscesses |
| 6 | Woman/26 | 1 | No | ANA 1:320, speckled | No | Asthma | Inflammatory | Oral | Subepidermal blister with abundant neutrophils and eosinophils |
| 7 | Woman/64 | 3 | Melanoma, breast adenocarcinoma, eccrine poroma | ANA 1:160, autoimmune optic neuritis | No | Osteoporosis, COPD, allergic rhinitis | Inflammatory | Oral, genital | Eosinophilic spongiosis |
| 8 | Woman/19 | 6 | No | ANA 1:1280, weak anti-ADN, anti-Ro+ | No | Arthritis | Inflammatory | Oral, esophageal | Subepidermal blister with abundant neutrophils |
| 9 | Woman/72 | 1 | No | No | No | Toxic hepatitis | Noninflammatory | Oral, conjunctival | Subepidermal blister with scant inflammatory response |
| Patient No. | Immunofluorescence | ELISA, Type VII Collagen | Immunoblotting | ||
| DIF | DIF, Salt-Split Human Skin | IIF, Salt-Split Human Skin | |||
| 1 | IgG, C3 | NP | Dermal, 1:320 | NP | Laminin 332 (IgG4) negative; type VII collagen (NC1 for IgG1, IgG3, IgG4) positive |
| 2 | IgG, IgM, C3 | Dermal | Negative | Negative | Anti-laminin 332 (IgG4) negative; type VII collagen (NC1 for IgG1, IgG3, IgG4) positive |
| 3 | IgG, IgA, C3 | NP | Dermal, IgA, and IgG 1:320 | Positive | NC1: IgG4 ++, IgA positive |
| 4 | IgG, C3 | Dermal | Negative | NP | Laminin 332 (IgG4) negative; collagen VII (NC1 for IgG1, IgG3, IgG4) positive |
| 5 | IgG, IgA, C3 | Dermal | Dermal, 1:160 | Negative | Laminin 332 (IgG4) negative; collagen VII (NC1 for IgG1, IgG3, IgG4) positive |
| 6 | IgG, IgA, C3 | Dermal | NP | NP | NP |
| 7 | IgG, C3 | Dermal | Dermal, 1:20 | Positive | NP |
| 8 | IgG, IgM, IgA, C3, fibrinogen | Dermal | Dermal, 1:20 (ANA dermal-epidermal) | Negative | NC1: IgG1-IgG4-IgA type VII collagen positive |
| 9 | IgG, C3 | NP | Dermal | NP | NP |
ABBREVIATIONS: ANA, antinuclear antibodies; COPD, chronic obstructive pulmonary disease; ELISA, ezyme-linked immunosorbent assay; HCV, hepatitis C virus; Ig, immunoglobulin; NIDDM, non-insulin-dependent diabetes mellitus; NP, not performed.
Malignant disease was a frequent association, with 5 tumors in 3 patients. Two tumors were in the lung, 1 was on the cervix, 1 in the breast, and 1 in the skin (melanoma). Other associations were inflammatory bowel disease (1 case of Crohn disease and 1 of ulcerative colitis) and autoimmune diseases or a finding of circulating antibodies (1 hemolytic anemia, 1 optic neuritis, 1 thyroiditis, 4 positive for antinuclear antibodies [ANA]) (Table 1).
Histology showed a subepidermal blister with scant inflammatory infiltrate for all 3 patients with classic EBA. In the 6 patients with inflammatory EBA, histology detected an inflammatory infiltrate with abundant neutrophils and eosinophils, and occasional microabscesses in the papillary dermis. Eosinophilic spongiosis was observed in 2 cases. DIF consistently showed linear IgG and C3 deposits of variable intensity along the basement membrane(Fig. 3). Other conjugates were detected in 5 patients (56%) (Table 1). IIF was performed with human salt-split skin and the serum samples from 8 patients; in 6 of these patients (75%) the result was positive and a dermal pattern was detected (Fig. 4). For 1 patient (case 6, Table 1), IIF performed in another center only detected the presence of ANA; serum was not available to us for IIF in salt-split human skin. Results of DIF on salt-split skin showed an exclusively dermal IgG deposition pattern for all 6 patients in whom this test was performed; this group included 2 patients with negative IIF results and a patient for whom IIF was not performed.

 of the blister. Original magnification ×200.")
Type VII collagen ELISA was performed in 5 cases and was positive in 2 (40%). Immunoblot analysis was performed in 6 cases and was positive in 5 (83%) (Fig. 5). These results are summarized in Table 1. ANA were detected in 4 patients (44%). In 1 case (patient 6, Table 1), this was an isolated finding. In 2 other cases (patients 3 and 7, Table 1) autoimmune diseases were also found. Although patient 8 (Table 1) was positive for ANA and anti-DNA, the ACR criteria for a diagnosis of BSLE were never met at any time during the course of disease. This patient was unresponsive to high doses of corticosteroids and dapsone but showed a good response to intravenous immunoglobulin (IVIG) therapy and colchicine. These outcomes suggested a diagnosis of EBA rather than BSLE even though the 2 processes have similar immunologic features and overlap to a certain extent.
. The 145-kDa band corresponds to the NC1 domain of type VII collagen. NC refers to negative control; PC, positive control; P, patient")
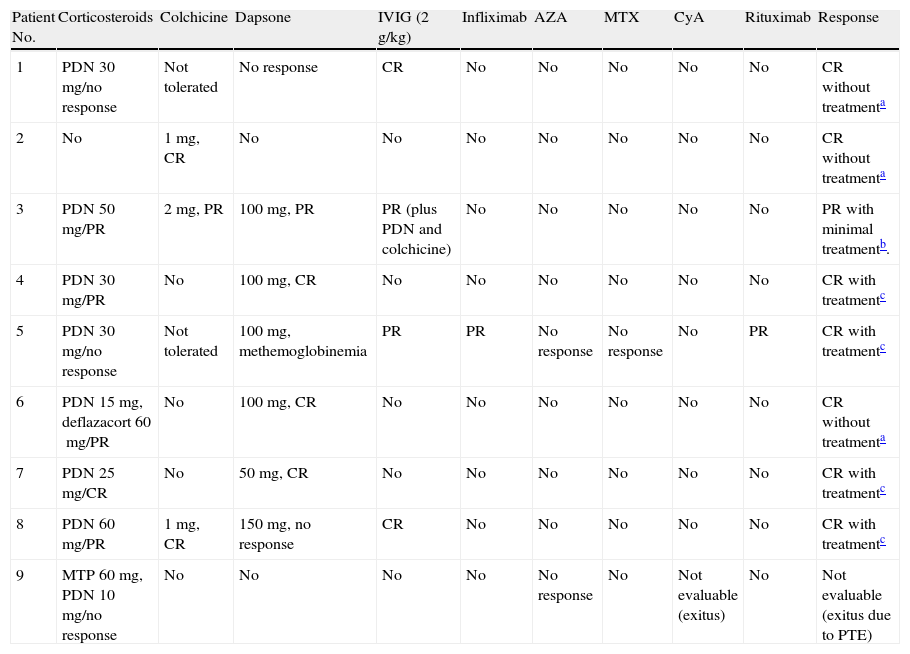
After diagnosis, a variety of treatments were tried; response was variable in most patients and disease was difficult to control in 2 cases (patients 3 and 5, Table 2). Patient 3 started treatment with prednisone at a daily dose of 1mg/kg, achieving partial response. Colchicine at a dose of 1mg every 12h was added, followed by dapsone (100mg/d), to little effect. High-dose IVIG therapy (2g/kg) was therefore started. There was still no response, but after prednisone and colchicine were added to the regimen, partial response was finally achieved. During the course of disease, pulmonary adenocarcinoma was detected; metastasis developed and the patient died. The case of patient 5 was the most refractory to therapy. Various treatments were tried and withdrawn, either because of adverse events or lack of response. In December 2007, rituximab was started (375mg/m2/wk for 4 weeks). There was good response to this regimen in the skin, but gingival erosion remained a problem. At the time of writing, the patient was in complete remission on low-dose corticosteroids, dapsone, and colchicine. We emphasize the excellent response to IVIG therapy in a patient with the classic form of EBA (patient 1, Table 2) because lesions had been refractory to all the treatments that had been tried previously.
Treatment and Response.
| Patient No. | Corticosteroids | Colchicine | Dapsone | IVIG (2g/kg) | Infliximab | AZA | MTX | CyA | Rituximab | Response |
| 1 | PDN 30mg/no response | Not tolerated | No response | CR | No | No | No | No | No | CR without treatmenta |
| 2 | No | 1mg, CR | No | No | No | No | No | No | No | CR without treatmenta |
| 3 | PDN 50mg/PR | 2mg, PR | 100mg, PR | PR (plus PDN and colchicine) | No | No | No | No | No | PR with minimal treatmentb. |
| 4 | PDN 30mg/PR | No | 100mg, CR | No | No | No | No | No | No | CR with treatmentc |
| 5 | PDN 30mg/no response | Not tolerated | 100mg, methemoglobinemia | PR | PR | No response | No response | No | PR | CR with treatmentc |
| 6 | PDN 15mg, deflazacort 60mg/PR | No | 100mg, CR | No | No | No | No | No | No | CR without treatmenta |
| 7 | PDN 25mg/CR | No | 50mg, CR | No | No | No | No | No | No | CR with treatmentc |
| 8 | PDN 60mg/PR | 1mg, CR | 150mg, no response | CR | No | No | No | No | No | CR with treatmentc |
| 9 | MTP 60mg, PDN 10mg/no response | No | No | No | No | No response | No | Not evaluable (exitus) | No | Not evaluable (exitus due to PTE) |
ABBREVIATIONS: AZA, azathioprine; CR, complete response; CyA, ciclosporin; IVIG, intravenous immunoglobulins; MTP, methylprednisolone; MTX, methotrexate; PDN, prednisone; PR, partial response; PTE, pulmonary thromboembolism.
EBA is an uncommon disease that mainly affects adults.1–3 The median age at diagnosis in our series was 37 years; women predominated, as in the 4 previously published studies.29–32
The classic mechanobullous form of EBA is the one that was first reported. This form has quite characteristic clinical features that facilitate diagnosis, undoubtedly explaining why most of the initial publications were about this phenotype. Since the inflammatory variety was described, more cases of this form have emerged. It is possible that greater understanding of EBA and appropriate use of the diagnostic techniques now available have enabled the diagnosis of cases that might previously have been thought to be bullous pemphigoid. The inflammatory form was more frequent in our series, as in other recently published ones. However the classic form predominated in our older patients, an observation that was not described in other series.29–32 We cannot explain this finding and the small number of patients precludes drawing conclusions. It is well known that EBA is often found in association with other diseases. The most clearly established association is probably with inflammatory bowel disease, particularly Crohn disease.1,3 The presence of type VII collagen in the intestinal epithelium and the phenomenon of epitope spreading could explain this association; this hypothesis is supported by the presence of circulating antibodies that target this collagen in approximately 68% of patients with Crohn disease even when no skin signs are present.33,34 In our series the incidence of tumors was higher in association with inflammatory bowel disease. Single cases of EBA associated with tumors have been reported, but the association has not been reported in any patient series until now. The course of disease in our patients did not seem to suggest a paraneoplastic process. In fact, the 3 tumors that had been diagnosed before the onset of EBA were in remission. In patient 3, diagnosis was delayed 72 months from onset and the tumors were detected at the same time; she did not receive immunosuppressants that might have triggered development of the tumor. In patient 2, the tumor presented after complete remission of EBA, which had been treated only with colchicine.
The degree of clinical inflammation correlated with the intensity of the inflammatory infiltrate, consistent with the literature on histologic findings in this disease. DIF did not allow us to differentiate EBA from bullous pemphigoid, even though we often observed that IgG deposits were more intense than C3 deposits in our EBA patients (in contrast with the more intense C3 deposition reported in pemphigoid35). The diagnostic method with the highest sensitivity was DIF with salt-split skin, which showed a dermal pattern in all cases where it was used. IIF with salt-split skin was negative in only 2 patients. The immune-deposit patterns revealed by DIF—either n-serrated or u-serrated—could not be assessed in our series, possibly because of technical problems related to the thickness of sections. We must point out that the results of the tests done in patient 5 did not lead to a definitive diagnosis of EBA. We cannot rule out that this slowly progressing case, which was refractory to treatment, was not IgA-mediated EBA because the appropriate ELISA was not done for this patient, who had associated Crohn disease and mucosal involvement and was negative for anti-BP 180 antibody. IgG deposition in the basement membrane, however, was much more intense than either IgA or C3 deposition.
Response to therapy was highly variable and not always satisfactory. We underline the excellent results obtained with IVIG infusion in a woman with the classic form of EBA who had not responded to several previous treatments (Table 2). The approach to EBA treatment we would suggest based on our experience would be to start prednisone (or an equivalent) at 0.5mg/kg in association with colchicine (1–2mg/d) and/or dapsone (25–100mg/d). If the patient responds well enough to allow the prednisone dose to be tapered to 5mg (or discontinued altogether), the corticosteroid therapy can be maintained. If the prednisone dose cannot be reduced (despite the addition of up to 100mg of dapsone and 2mg of colchicine) we suggest adding IVIG therapy. Rituximab currently seems to be more effective at a lower cost, and this drug could be a good choice in cases that are refractory to other treatments. However, our experience with rituximab in EBA is limited to a single patient.
In summary, we present the first Spanish series of EBA patients. Consistent with the only other 4 series published29–32 (analyzing 38, 30, 14, and 12 patients, respectively), we found that inflammatory EBA predominated. In EBA, clinical suspicion must be high and optimal use of available diagnostic tools will be necessary for reaching a firm diagnosis. Lehman and colleagues36 proposed a diagnostic algorithm for autoimmune bullous diseases that took into consideration the cost of the various tests so that they can be used rationally. After diagnosis, the extent of disease and the possibility of associated conditions must be carefully assessed, and appropriate treatment should be started so that involvement of the eye and mucosae of the respiratory and digestive tracts can be prevented. Because this disease is highly variable in presentation, it is difficult to evaluate response to a particular drug.
This retrospective study included a small number of patients. Prospective multicenter studies are needed to improve our understanding of the mechanisms involved in EBA, to assess the efficacy of the drug treatments available, and to provide a basis for tailoring treatment.
Ethical DisclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that they have followed the protocols of their hospitals concerning the publication of patient data, and that all the patients included in this study were appropriately informed and gave their written informed consent.
Right to privacy and informed consentThe authors declare that no private patient data are disclosed in this article.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
We thank Dr C. Sitaru, who performed the type VII collagen ELISA assays, and Drs J. Herrero and A. Guilabert, who performed the immunoblotting analyses.
Please cite this article as: Barreiro-Capurro A, Mascaró-Galy JM, Iranzo P. Estudio retrospectivo de las características clínicas, histológicas e inmunológicas en una serie de de 9 pacientes con epidermólisis ampollosa adquirida. Actas Dermosifiliogr. 2013;104:904–914.


