




Inflammatory myxohyaline tumor of the distal extremities is an extremely rare low-grade sarcoma with a tendency to produce local recurrence after surgical excision, but with a low metastatic potential. We present the case of a 49-year-old woman with a slow-growing asymptomatic tumor on the right pretibial region that was initially considered to be a lipoma. Histopathology revealed the presence of a polymorphic inflammatory infiltrate within a myxoid and hyaline matrix. Interspersed between the inflammatory cells were 3 different populations of neoplastic cells: atypical spindle-shaped cells; bizarre epithelioid cells, some of which were multinucleated and resembled the virocytes or Reed-Sternberg cells; and cells with abundant, vacuolated cytoplasm, similar to lipoblasts. These clinical-pathologic findings led to a diagnosis of inflammatory myxohyaline tumor of the distal extremities. Although the tumor was excised with wide surgical margins, local recurrence developed after 3 months and was treated with re-excision and radiotherapy.
El tumor mixohialino inflamatorio de las áreas distales de las extremidades (TMHIADE) es un sarcoma de bajo grado de malignidad extremadamente infrecuente, con tendencia a la recurrencia local tras su extirpación quirúrgica, pero con un bajo potencial metastásico. Presentamos el caso de una mujer de 49 años que consultó por una tumoración asintomática de lento crecimiento en la zona pretibial derecha, que inicialmente sugirió el diagnóstico de lipoma. El estudio histopatológico mostró la presencia de un infiltrado inflamatorio polimórfico inmerso en una matriz mixoide e hialina. Entremezcladas entre las células inflamatorias existían varias poblaciones de células tumorales: en primer lugar, unas células atípicas de morfología fusiformes; en segundo lugar, unas células epitelioides bizarras, algunas de las cuales eran multinucleadas y recordaban a los virocitos o células de Reed-Sternberg; por último, unas células de citoplasma amplio multivacuolado que recordaban a los lipoblastos.
Estos hallazgos clínico-patológicos permitieron el diagnóstico de tumor mixohialino inflamatorio de las áreas distales de las extremidades. A pesar de que la tumoración se extirpó con márgenes quirúrgicos amplios, presentó una recidiva local tres meses después que fue tratada con nueva extirpación quirúrgica y radioterapia.
The diagnosis of subcutaneous tumors can often pose significant problems in daily clinical practice, leading to difficulties when taking therapeutic decisions. We describe a patient with a subcutaneous mass that was finally diagnosed as an inflammatory myxohyaline tumor of distal extremities (IMTDE), a very rare tumor with a complex but characteristic histological pattern.
Case DescriptionA 49-year-old woman consulted for an asymptomatic subcutaneous mass that had developed 2 months earlier in the pretibial region of the right leg. She reported no history of trauma or surgery to the area. The mass had grown progressively over the previous weeks and gave rise to mild discomfort. Physical examination revealed a well-defined, skin-colored nodule that was firm and elastic on palpation (Fig. 1A). The nodule measured 5x6x2cm (Fig. 1B), and the diagnoses considered were cutaneous lipoma or a malignant soft-tissue tumor. Blood tests including complete blood count, electrolytes, and biochemistry were normal, and imaging studies revealed a noncalcified subcutaneous soft-tissue mass that did not appear to affect the underlying muscle fascia. A deep incisional biopsy was performed and histological examination after staining with hematoxylin and eosin showed the tumor to be situated in the dermis and subcutaneous tissue and to be formed of a relatively sparse inflammatory infiltrate of lymphocytes, neutrophils, plasma cells, histiocytes, and numerous eosinophils in a myxoid and hyaline stroma (Fig. 2A). There were 3 populations of tumor cells. One was composed of cells with an epithelioid appearance, some of them with abundant basophilic cytoplasm and bizarre hyperchromatic nuclei with marked nuclear pleomorphism, and others with multiple vesicular nuclei and prominent eosinophilic nucleoli, similar to virocytes or Reed-Sternberg cells (Fig. 2B). The second cell population was of spindle-shaped neoplastic cells with basophilic cytoplasm and atypical nuclei (Fig. 2C). The third population was formed of cells similar to lipoblasts, with abundant cytoplasm containing numerous vacuoles (Fig. 2D). Immunohistochemistry showed that the 3 tumor-cell populations were strongly positive for vimentin (Fig. 3A), moderately positive for CD68 (Fig. 3B) and CD34 (Fig. 3C), and negative for S-100, CD45 (leukocyte common antigen), CD15, CD30, CD31, cytokeratin CAM5.2, desmin, and α-actin. Although there was marked cell atypia, the number of mitoses was moderate (Ki-67 proliferation index, 30%) (Fig. 3D). A diagnosis of IMTDE was made on the basis of the clinical, histopathological, and immunohistochemical findings, and wide excision of the tumor was therefore performed with margins greater than 2cm and inclusion of the periosteum; the defect was closed with a skin graft. The macroscopic appearance of the surgical specimen was of a multinodular tumor with areas of hemorrhage; the tumor infiltrated the hypodermis, with nodules present down to the periosteum (Fig. 1B). The surgical margins were free of disease, but tumor deposits were detected in the resected periosteum. The tumor recurred 3 months after the operation (Fig. 4); this was treated by further surgical excision and grafting and a cycle of postoperative radiation therapy. At the time of writing, after 9 months of follow-up, the patient was healthy and disease free.

A, In the right pretibial region, a poorly defined subcutaneous mass that had shown rapid growth in the previous weeks. The initial diagnosis was lipoma. B, Macroscopic image showing a poorly delimited multinodular subcutaneous tumor of 5x6.5x2cm. The tumor had a gelatinous appearance and there were areas of hemorrhage. Nodules infiltrating the adipose tissue and the periosteal fat were observed.
. B, Population of tumor cells formed of multinucleated Reed-Sternberg-like cells and epithelioid cells (Hematoxylin-eosin, original magnification x400). C, Spindle-shaped tumor cells (Hematoxylin-eosin, original magnification x400). D, Multivacuolated lipoblast-like cells (Hematoxylin-eosin, original magnification x400).")
A, Foci of dense polymorphic inflammatory infiltrate. Lymphocytes, plasma cells, neutrophils, and numerous eosinophils are present within the myxoid matrix, forming part of the tumor (Hematoxylin-eosin, original magnification x200). B, Population of tumor cells formed of multinucleated Reed-Sternberg-like cells and epithelioid cells (Hematoxylin-eosin, original magnification x400). C, Spindle-shaped tumor cells (Hematoxylin-eosin, original magnification x400). D, Multivacuolated lipoblast-like cells (Hematoxylin-eosin, original magnification x400).


IMTDE, also known as acral myxoinflammatory fibroblastic sarcoma or inflammatory myxohyaline tumor of distal extremities with virocytes or Reed-Sternberg-like cells, was first described by Montgomery in 1997.1
This low-grade sarcoma is extremely rare and shows a tendency to local recurrence, particularly after incomplete surgical excision, though it has a low metastatic potential.1,2 The incidence is similar in men and women, with a peak between 30 and 50 years. The majority of patients present with a poorly defined, painless mass in the distal regions of the limbs, and the mean duration of symptoms prior to presentation is 1 year.3 Tumor growth is progressive, although tumors that grow rapidly over a few weeks have been described. The upper limbs are affected more frequently than the lower limbs, the fingers and hands being the most common sites, followed by the arms. In the lower limbs, the tumor occurs with highest frequency on the toes and dorsum of the foot, followed by the distal regions of the legs. Some patients report local discomfort or mild functional disability, particularly when tumor growth is rapid.3,4 The clinical diagnosis prior to biopsy is usually of a benign lesion of inflammatory origin (60%),1 such as tenosynovitis, or of a tumor, one of the most common clinical diagnoses being lipoma, as occurred in our patient. Magnetic resonance imaging is a very useful diagnostic tool; the findings are consistent with a poorly defined, invasive soft-tissue tumor of the subcutaneous tissue, with spread into the underlying adipose tissue and into nearby tendons.
IMTDE is a multinodular tumor with poorly defined borders, meaning that the initial excision is often incomplete. The mean diameter is 4cm (range, 1-9cm).3
Macroscopically it has areas of gelatinous appearance, due to an abundance of mucin, and areas of hemorrhage, as were observed in our case. A large percentage of these tumors are associated with synovitis. The most characteristic finding on histology is the presence of a marked inflammatory component formed of neutrophils, lymphocytes, plasma cells, and eosinophils that extends diffusely through the myxoid and hyaline areas of the tumor, around a very rich vascular network, giving the appearance of granulation tissue.4 The ratio of myxoid to hyaline areas is variable, sometimes even within a single tumor. In some cases the tumor is formed principally of myxoid tissue with few cells, whereas, in others, the mucin is present in more focal deposits.5 In the myxohyaline areas the tumor cells may be interspersed with the inflammatory cells. In areas of higher tumor density, the neoplastic cells may be divided into 3 populations of different morphology: the first type is of bizarre spindle-shaped cells with basophilic cytoplasm and atypical nuclei, as were observed in our case. The second type is of large, polygonal, ganglion-type epithelioid cells with basophilic cytoplasm and oval nuclei with marked nuclear atypia, vesicular chromatin, and prominent, large nucleoli1; some of these cells are multinucleated, similar to the virocytes or Reed-Sternberg cells that may be found in Hodgkin disease, and that were also present in our patient. These giant cells occasionally present emperipolesis and may contain lymphocytes, eosinophils, and neutrophils, or even plasma cells and red blood cells1,6–8; emperipolesis was not observed in the tumor from our patient. The third population is of multivacuolated cells of variable size and with abundant eosinophilic cytoplasm; these cells are similar to pleomorphic lipoblasts and were also detected in the histological study of the tumor from our patient. Mitoses do not usually exceed 1 per high-power field, with a proliferation index of less than 5% in over 90% of cases,1 and atypical mitotic figures are not seen. Necrosis is not a common feature of this tumor. Immunohistochemistry of IMTDE shows that the neoplastic cells are diffusely positive for vimentin, with occasional CD68+ multinucleated cells. Some of the cells may be positive for CD34. Other markers, such as cytokeratin CAM5.2, keratin AE1/AE3, epithelial membrane antigen, protein S100, HMB-45, melan-A, smooth muscle actin, desmin, factor VIII, CD15, CD30, leukocyte common antigen, and CD15 (Leu-M1), are negative.1,6,8,9
Diagnosis of IMTDE is complex due to the peculiar histopathological characteristics of this sarcoma. A biopsy that is too superficial can lead to diagnostic errors arising from confusion with a benign myxoid tumor or an inflammatory lesion. Prior to its definitive description in 1997, IMTDE was often incorrectly diagnosed as Hodgkin disease or soft-tissue sarcoma not otherwise specified; it has even been confused with inflammatory or infectious disorders due to the presence of a typically dense polymorphous inflammatory infiltrate, which has also been described in other types of sarcoma but which is particularly prominent in IMTDE.1,2,4 In addition, neoplastic cells may be scarce and distributed diffusely, causing them to be hidden by the dense inflammatory infiltrate and leading to erroneous diagnosis as a reactive disorder or inflammatory pseudotumor.1–3,5 A further problem with a superficial biopsy is that it will not allow us to observe infiltration of the adipose tissue, tendons, or periosteum by the neoplastic cells; this type of infiltration is very common in IMTDE and rare in inflammatory conditions. For this reason, deep biopsy is always recommended when a soft-tissue tumor is suspected. One of the conditions that must be considered in the differential diagnosis of IMTDE is Rosai-Dorfman disease, a reactive condition that also presents emperipolesis, though without the cytoplasmic or nuclear vacuoles that are observed in the neoplastic cells of IMTDE; immunohistochemistry is positive for S-100 in Rosai-Dorfman disease whereas it is negative in IMTDE.1,2,8
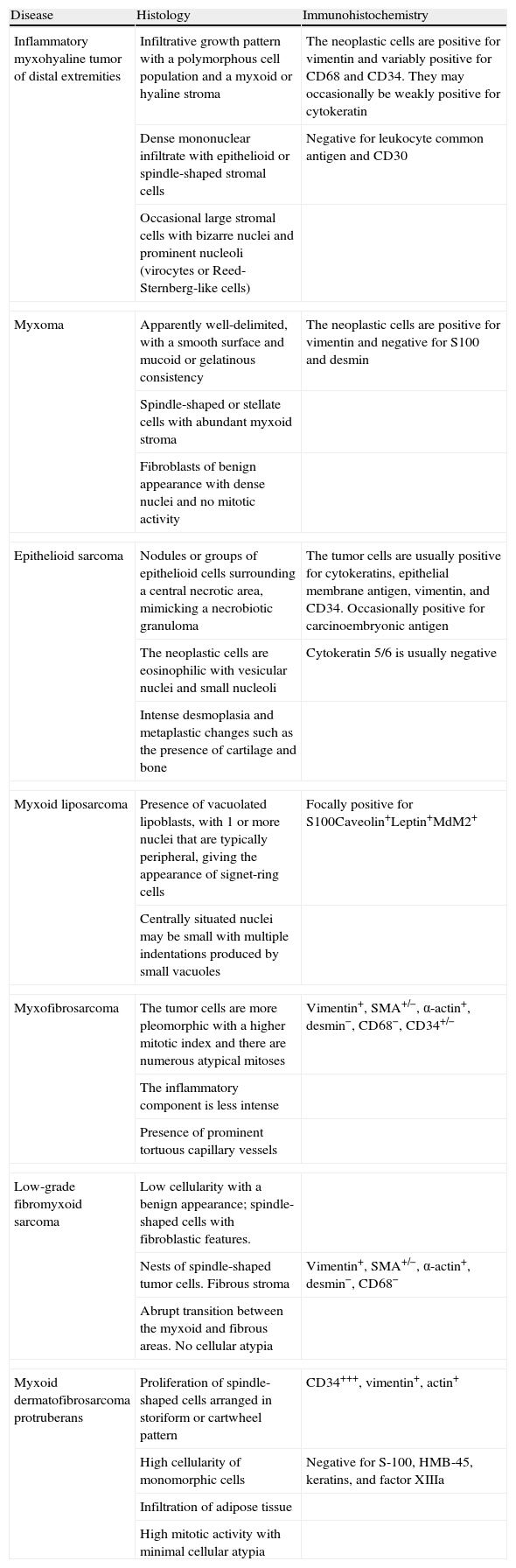
The differential diagnosis of IMTDE is extensive and depends on which component predominates in the tumor: myxoid, inflammatory, or bizarre atypical cells. First, other benign and malignant soft-tissue myxoid tumors must be excluded, the most important of which is the myxoid variant of low-grade malignant fibrous histiocytoma (myxofibrosarcoma). This tumor usually affects older individuals of 50 to 80 years of age and typically appears on the proximal regions of the limbs, in contrast to the distal location of IMTDE. The histological findings include more marked cell pleomorphism in myxofibrosarcoma than in IMTDE, as well as a much higher mitotic index and a large number of atypical mitoses.9 In addition, although myxofibrosarcoma can present an associated inflammatory reaction, it is usually less intense than in IMTDE and there is an absence of emperipolesis. Immunohistochemistry of myxofibrosarcoma shows the cells to be weakly positive for vimentin; some cells express smooth muscle actin and α-actin and, in contrast to the situation in IMTDE, the tumor is usually negative for desmin, CD68, and CD34. Myxofibrosarcoma carries a poorer prognosis than IMTDE as distant metastases are common (Table 1).1–4,8,9 Other malignant tumors with a myxoid stroma that must be distinguished from IMTDE are myxoid liposarcoma, low-grade fibromyxoid sarcoma, malignant myxoid peripheral nerve sheath tumor, the myxoid variant of dermatofibrosarcoma protruberans (Table 1), and extraskeletal myxoid chondrosarcoma.
Differential Diagnosis of Inflammatory Myxohyaline Tumor of Distal Extremities.
| Disease | Histology | Immunohistochemistry |
| Inflammatory myxohyaline tumor of distal extremities | Infiltrative growth pattern with a polymorphous cell population and a myxoid or hyaline stroma | The neoplastic cells are positive for vimentin and variably positive for CD68 and CD34. They may occasionally be weakly positive for cytokeratin |
| Dense mononuclear infiltrate with epithelioid or spindle-shaped stromal cells | Negative for leukocyte common antigen and CD30 | |
| Occasional large stromal cells with bizarre nuclei and prominent nucleoli (virocytes or Reed-Sternberg-like cells) | ||
| Myxoma | Apparently well-delimited, with a smooth surface and mucoid or gelatinous consistency | The neoplastic cells are positive for vimentin and negative for S100 and desmin |
| Spindle-shaped or stellate cells with abundant myxoid stroma | ||
| Fibroblasts of benign appearance with dense nuclei and no mitotic activity | ||
| Epithelioid sarcoma | Nodules or groups of epithelioid cells surrounding a central necrotic area, mimicking a necrobiotic granuloma | The tumor cells are usually positive for cytokeratins, epithelial membrane antigen, vimentin, and CD34. Occasionally positive for carcinoembryonic antigen |
| The neoplastic cells are eosinophilic with vesicular nuclei and small nucleoli | Cytokeratin 5/6 is usually negative | |
| Intense desmoplasia and metaplastic changes such as the presence of cartilage and bone | ||
| Myxoid liposarcoma | Presence of vacuolated lipoblasts, with 1 or more nuclei that are typically peripheral, giving the appearance of signet-ring cells | Focally positive for S100Caveolin+Leptin+MdM2+ |
| Centrally situated nuclei may be small with multiple indentations produced by small vacuoles | ||
| Myxofibrosarcoma | The tumor cells are more pleomorphic with a higher mitotic index and there are numerous atypical mitoses | Vimentin+, SMA+/−, α-actin+, desmin−, CD68−, CD34+/− |
| The inflammatory component is less intense | ||
| Presence of prominent tortuous capillary vessels | ||
| Low-grade fibromyxoid sarcoma | Low cellularity with a benign appearance; spindle-shaped cells with fibroblastic features. | |
| Nests of spindle-shaped tumor cells. Fibrous stroma | Vimentin+, SMA+/−, α-actin+, desmin−, CD68− | |
| Abrupt transition between the myxoid and fibrous areas. No cellular atypia | ||
| Myxoid dermatofibrosarcoma protruberans | Proliferation of spindle-shaped cells arranged in storiform or cartwheel pattern | CD34+++, vimentin+, actin+ |
| High cellularity of monomorphic cells | Negative for S-100, HMB-45, keratins, and factor XIIIa | |
| Infiltration of adipose tissue | ||
| High mitotic activity with minimal cellular atypia | ||
Abbreviations: HMB: human melanoma black; MdM: murine double minute; S-100: protein S-100; SMA, smooth muscle actin.
Another tumor that must be included in the differential diagnosis is epithelioid sarcoma, which also appears on distal areas of the body in young and middle-aged patients. The observation of Reed-Sternberg-like cells raises the possibility of Hodgkin disease, although the presence of the other neoplastic-cell populations characteristic of IMTDE exclude this diagnosis.
IMTDE must also be distinguished from certain benign soft-tissue tumors, especially myxoma (Table 1). This diagnosis is most likely to be suspected in cases of IMTDE with low levels of cellularity and a very large myxoid component, particularly when the biopsy is superficial and does not enable us to detect the malignant cells of IMTDE. Other benign tumors to be considered in the differential diagnosis are myxoid lipoma, digital mucous cyst, and myxoid neurofibroma.
Ultrastructural studies have shown that the neoplastic cells of IMTDE present features of modified fibroblasts, including abundant intermediate filaments and dilated rough endoplasmic reticulum. It has not been possible to demonstrate the constant presence of associated cytogenetic abnormalities in this sarcoma, though the following have been reported: translocation t(1;10)(p22;q24),10 dicentric chromosomes secondary to the rupture of chromosomes 7 and 8, translocation t(2;6)(q31;p21.3),11 losses from chromosomes 3 and 13, and genetic gain due to the addition of genetic material to the short arm of chromosome 13.12
IMTDE has a tendency to local recurrence, particularly if surgical excision is incomplete, as occurred in our case. The majority of recurrences are multiple and aggressive, and amputation of the limb is required in up to 30% of cases. Metastases to distant lymph nodes and to the lungs have been reported occasionally (2%-6% at 5 years). The treatment of choice is wide surgical excision, aiming to achieve disease-free margins. If recurrence develops, reoperation is recommended. The adjuvant treatments proposed for use in unresectable or recurrent tumors include radiation therapy and epithelial growth factor receptor (EGFR) inhibitors, particularly in those tumors in which the cells are positive for EGFR.7,9,13
In conclusion, we present a case of IMTDE, a rare condition that must be recognized due to its frequent clinical confusion with benign disorders, the broad differential diagnosis, and the difficulties of correct histopathological diagnosis.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Agusti-Mejias A, et al. Tumor mixohialino inflamatorio de las zonas distales de las entremidades. Actas Dermosifiliograf. 2011;102:456-462.



