
INTRODUCCION
Las neoformaciones vasculares (hemangiomas) pueden formar parte de distintos síndromes malformativos, principalmente neurológicos y óseos; entre estos últimos se encuentra el síndrome de Maffucci, descrito por este autor en 18811 y que asocia la presencia, generalmente de aparición simultánea, de hemangiomas y encondromas. La importancia del cuadro no sólo radica en su aspecto puramente malformativo, sino en la producción de defectos óseos y en la asociación, por lo general alta, a lesiones tumorales benignas y malignas.
DESCRIPCION DEL CASO
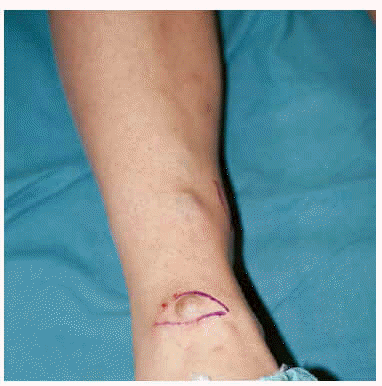
Mujer de 26 años que desde la primera infancia ha presentado tumores subcutáneos blandos que han tendido a aumentar de tamaño y volverse dolorosos con el paso del tiempo. Estas neoformaciones se localizaban en ambos miembros inferiores, así como en un miembro superior y zona derecha de tórax. El tamaño de los elementos oscilaba entre los 0,5 y 5 cm de diámetro, no alteraban el color de la piel suprayacente salvo en las zonas distales de los miembros inferiores donde se apreciaba el color azulado de la neofor-mación vascular subyacente (fig. 1). El número de los mismos ha tendido a aumentar, alcanzando en el momento actual el de 30. En general se apreciaba un aumento de partes blandas, deformidades óseas y acortamiento de los miembros. Asimismo, la paciente refería haber sufrido fracturas óseas en varias ocasiones.
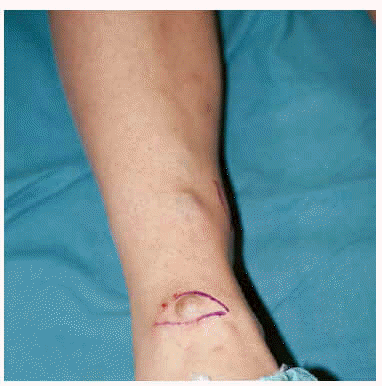
Fig. 1.--Lesiones tumorales subcutáneas en piernas.
Debido a la existencia de dolores óseos había sido diagnosticada en 1983 de encondromatosis múltiple, presentando afectación de rodilla izquierda con importante genu valgo, por el que había sido intervenida en dos ocasiones, así como afectación de hombro derecho y evidente escoliosis por dismetría pélvica que compensaba mediante calza y tratamiento con cinesiterapia.
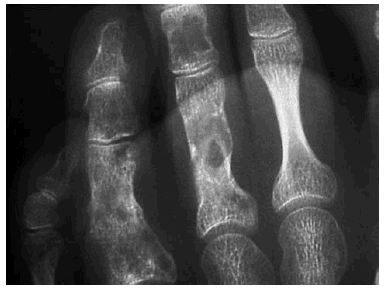
En el estudio radiológico realizado se observaban, junto a secuelas de antiguas fracturas, múltiples lesiones constituidas por imágenes radiolúcidas, a veces con borde escleroso o desflecado, localizadas a nivel de la parrilla costal, húmero, cúbito, radio, sacro, fémur, tibia, peroné, así como huesos de tarso, carpo y falanges. Dichas lesiones eran sugestivas de encondromatosis múltiple (fig. 2) que en algunos lugares coincidían con la localización de los hemangiomas.
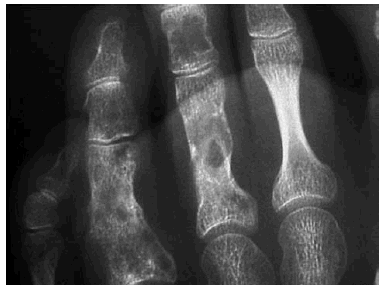
Fig. 2.--Encondromas óseos.
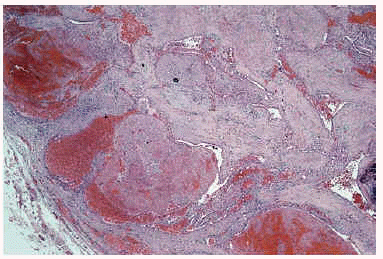
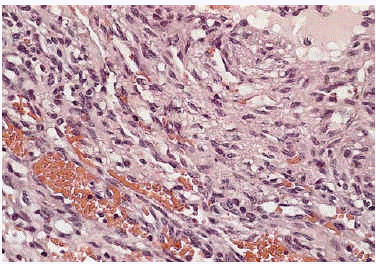
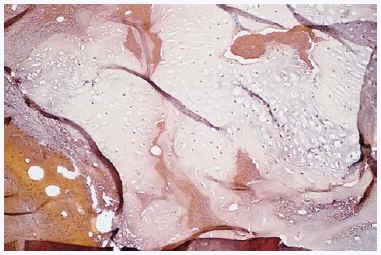
Algunas de las tumoraciones blandas, las que producían dolor, se extirparon y se realizó estudio histopatológico que demostró la presencia de espacios anfractuosos tapizados por endotelio y a menudo ocupados por trombos y hemorragias (fig. 3). En el abundante tejido interpuesto, además de fibrosis hay haces de células fusiformes con vacuolización citoplasmática (fig. 4) que caracteriza la entidad conocida como hemangioendotelioma, o actualmente hemangioma de células fusiformes.
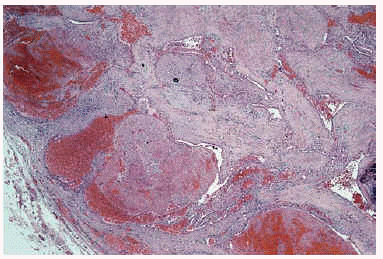
Fig. 3.--Lesión vascular profunda que afecta hasta hipodermis.
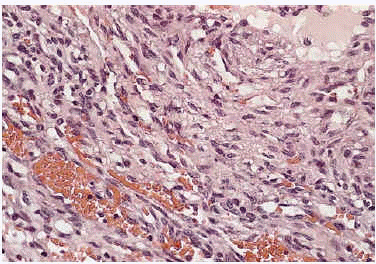
Fig. 4.--Detalle de las células fusiformes con espacios anfractuosos tapizados por endotelio y ocupados por trombos y hemorragia situadas entre las luces.
El estudio histológico de las lesiones óseas confirmó el diagnóstico radiológico por la presencia de tejido cartilaginoso maduro en el interior del hueso (fig. 5); no se hallaron signos de malignidad.
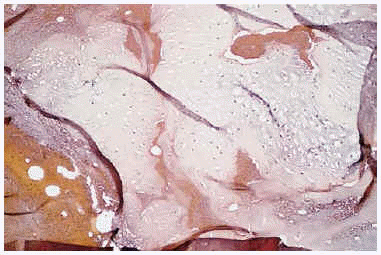
Fig. 5.--Nido de cartílago maduro en el espesor del hueso correspondientes a un encondroma.
En el seguimiento realizado a la paciente tanto clínico, analítico como radiológico no se ha demostrado hasta el momento la presencia de alteraciones sugestivas de malignidad.
DISCUSION
El síndrome de Maffucci, también conocido síndrome de discondrodisplasia con hemagiomas, o síndrome de Kast-Maffucci, corresponde a una displasia mesodérmica congénita no hereditaria caracterizada por la presencia de hemangiomas, encondromas y tendencia al desarrollo de lesiones degenerativas benignas y/o malignas.
Es un proceso poco frecuente. Lewis et al2 en la revisión retrospectiva que realizaron en 1973 sólo se encontraron 105 casos en la literatura anglosajona, a los que habría que añadir los 63 revisados por Kaplan et al en 1993. Sólo hemos encontrado dos casos publicados en la literatura dermatológica española4, 5; no hay diferencias de sexo o raza.
Es una enfermedad congénita a la que no se considera hereditaria, aunque pueden demostrarse antecedentes en el 27% de los casos. La edad de comienzo es en torno a los 4 años3. Aunque Matsumoto et al6 han observado alteraciones cromosómicas, estos hallazgos no han sido corroborados en estudios posteriores.
Las alteraciones óseas descritas en el síndrome son debidas a la presencia de encondromas, tumores que se caracterizan por la persistencia ectópica de cartílago en las metáfisis y diáfisis de los huesos, lo que provoca su deformidad y acortamiento como consecuencia de su expansión en el interior de los mismos. Son causa, por tanto, de deformidades y fracturas espontáneas. La escoliosis está presente en 17% de los pacientes, el acortamiento óseo en el 15% y la fractura espontánea en el 20%-26% de los casos publicados3, 7.
Se han descrito 5 patrones de encondromas: acral, regional, unilateral, multifocal y generalizado, pudiendo, como en nuestro caso, existir patrones mixtos. No es raro la coexistencia en la misma región de alteraciones vasculares y óseas4, que también podían apreciarse en nuestra paciente.
Las lesiones vasculares suelen corresponder a hemangiomas cavernosos, si bien se han descrito neoformaciones capilares linfáticas, pero muy especialmente hemangioendotelioma de células fusiformes2, 3, 8, 9, ya que la mayoría de los pacientes con este síndrome presentan este tipo de lesión vascular hoy denominado hemangioma de células fusiformes10, 11. Clínicamente se caracterizan por la presencia de tumores subcutáneos que pueden dar a la piel un color azulado, son blandos y desaparecen con la presión o con la elevación del miembro. Su número puede ser muy variable y tener una distribución uni o bilateral, habiéndose descrito afectación visceral: leptomeninges, ojos, faringe, intestino, lengua, tráquea y huesos12-16, lo que justifica la realización de técnicas de diagnóstico de imagen como la tomografía axial computarizada o la resonancia nuclear magnética.
En una ocasión se ha descrito coexistencia de hemangiomas con linfagiomas17.
Aunque la presencia de fracturas espontáneas y deformaciones óseas ya le dan trascendencia al síndrome, éste se ve agravado por la tendencia a la asociación de tumores malignos, ya referida desde la primera descripción de Maffucci. Los porcentajes de malignización han sido variables según las distintas series estudiadas; así, Lewis y Ketchman2 lo encontraron en el 23% y Kaplan et al3 en el 30%; mucho más pesimista es la opinión de Schawartz et al, que piensan que «con el tiempo la presencia de lesiones malignas se acerca al 100%18». El tumor maligno más frecuentemente descrito es el condrosarcoma, que aparece hasta el 37% de los pacientes3 y generalmente lo hace sobre lesiones previas de encondroma, aunque también pueden presentarse como tumores primitivos. Su edad de aparición es muy amplia, habiéndose descrito casos entre el espectro de los 13 y 69 años, estableciéndose la media en los 40 años2; el signo más evocador es el de la aparición de dolor, por lo que es recomendable proceder a la realización de estudios radiológicos y biopsia y extirpación si el enfermo refiere este síntoma. Se han publicado otra serie de tumores asociados al síndrome de Maffucci como astro-citoma, adenocarcinoma pancreático, hepático y biliar, fibrosarcoma ovárico y leucemia mieloide crónica, así como degeneración de los propios tumores vasculares (hemangiosarcoma y hemolinfangiosarcoma)2, 3, 19. Un hecho habitual es que los pacientes con síndrome de Maffucci puedan desarrollar más de un tumor maligno, siempre de origen mesodérmico; de hecho Kaplan et al3 encontraron 49 tumores en 31 enfermos. Por todo ello es necesario incluir el síndrome de Maffucci entre los síndromes con tendencia a la producción de tumores malignos.
Aunque el diagnóstico diferencial de síndrome de Maffucci no crea en principio muchas dificultades, habría que hacerlo con otras angiomatosis como el blue rubber bleb nevus, el síndrome de Klippel-Trenaunay, el síndrome de Gorham e incluso el sarcoma de Kaposi; sin embargo, las mayores dificultades las plantea con el síndrome de Ollier, o discondroplasia de Ollier, caracterizado por la presencia de encondromas, semejantes a los del Maffucci, y que como ellos pueden degenerar a condrosarcomas. En definitiva, ambos síndromes comparten las mismas características a excepción de los hemangiomas, lo que obliga a la realización de estudios profundos para establecer uno u otro diagnóstico, a pesar de lo cual no siempre logra establecerse20.
La poca frecuencia de este síndrome, como comentan Roson et al4, hace que existan muchas incógnitas sobre el mismo, de ahí el interés de comunicar todos los casos encontrados y tratar de agruparlos en series amplias para llegar a conocerlo mejor.



