

Introducción
El síndrome de Sweet (SS) o dermatosis aguda, febril, neutrofílica, fue descrito en 1964 como una erupción de placas y/o nódulos dolorosos, caracterizados histológicamente por un denso infiltrado dérmico compuesto de forma fundamental por neutrófilos1.
Los criterios diagnósticos del SS fueron propuestos por Su y Liu en 19862 y revisados posteriormente por Von der Driesch en 19893 y 19944.
Cuando el SS aparece en el contexto de una enfermedad maligna, tanto si precede a su diagnóstico como en los casos en que aparece durante el desarrollo de la misma, suele ser un marcador de mal pronóstico5. Sin embargo, la dermatosis en sí misma habitualmente es un cuadro agudo, recortado y de comportamiento benigno que suele presentar una buena respuesta al tratamiento esteroideo. Raramente se han descrito casos en que el SS tiene una evolución fatal6-13.
Una respuesta inflamatoria intacta es esencial para el mantenimiento de los mecanismos de defensa normales del huésped. Sin embargo, en algunos casos, esta respuesta puede ser muy intensa y causar un síndrome de disfunción orgánica múltiple. Este hecho es conocido como síndrome de respuesta inflamatoria sistémica en el cual se produce una secreción masiva de citocinas proinflamatorias como interleucina 6 (IL-6) y factor de necrosis tumoral alfa (TNF-alfa). Cuando el estímulo antigénico es mantenido, se produce un síndrome de respuesta inflamatoria crónico con deterioro progresivo del estado general y del estado inmunológico del paciente14,15.
La asociación del SS a un síndrome de respuesta inflamatoria sistémica se ha descrito raramente16-18. Presentamos un caso de SS de evolución crónica y recurrente asociado a un síndrome de respuesta inflamatoria sistémica idiopática que llevó a la muerte del paciente tras 7 años de evolución.
Caso clínico
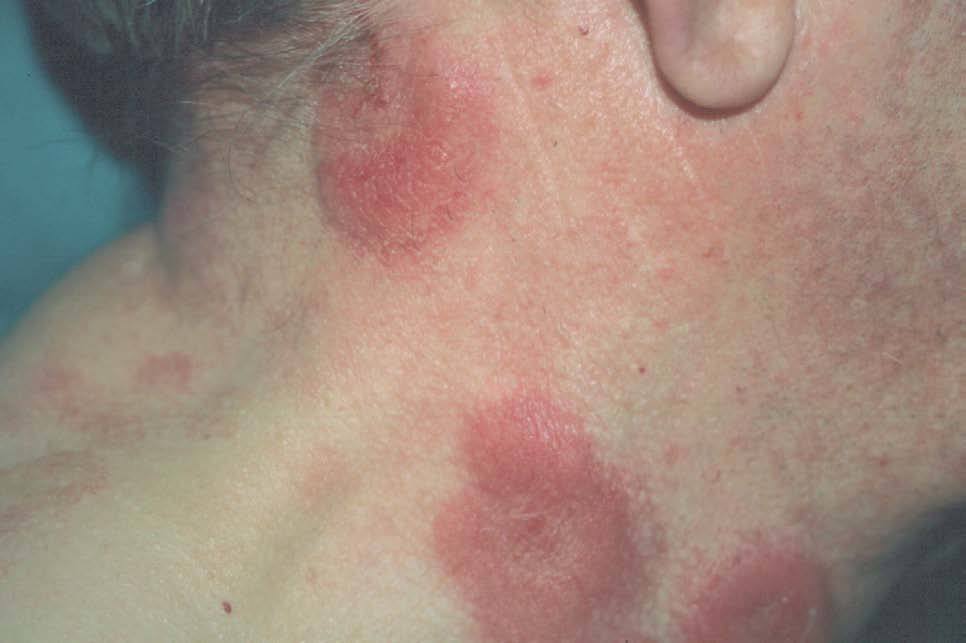
Un paciente varón de 72 años fue valorado en agosto de 1998 por presentar masas eritematosas pseudotumorales en cara anterior del tronco y región facial (fig. 1), acompañadas de fiebre y malestar general. El paciente refería presentar de forma episódica brotes de lesiones similares durante los últimos 3 años. Ya se había realizado una biopsia en otro centro informada como dermatosis neutrofílica, y los estudios analíticos, radiológicos, serológicos y marcadores tumorales habían sido normales o negativos.
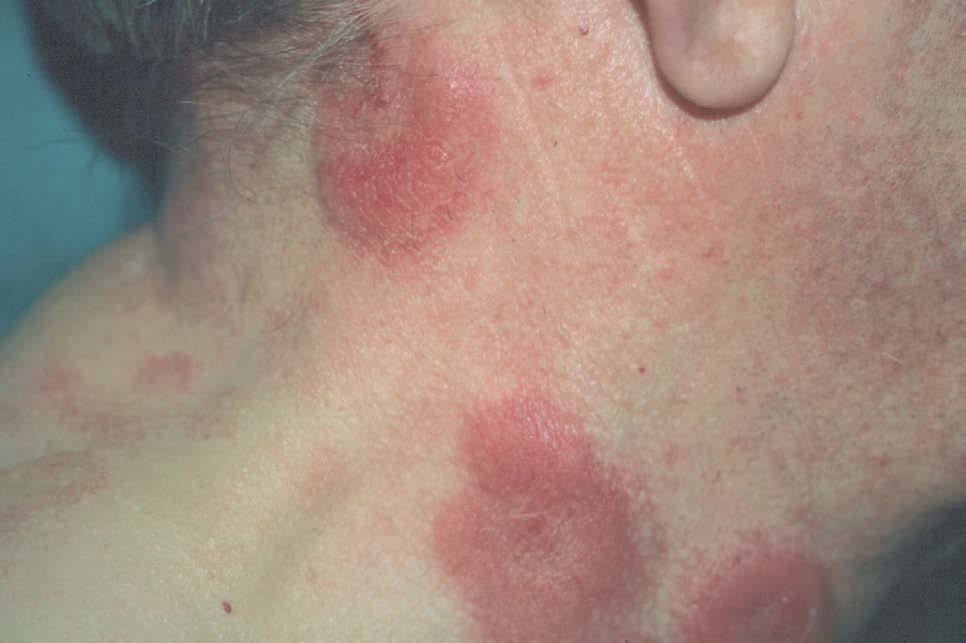
Figura 1. Masas pseudotumorales eritemato-edematosas localizadas en cuello.
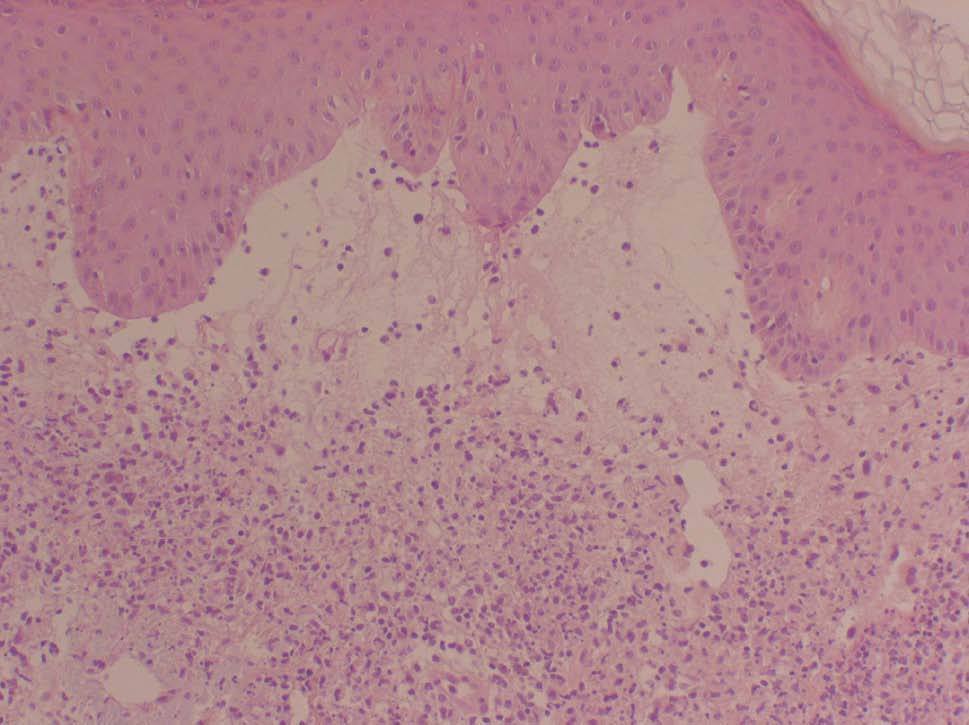
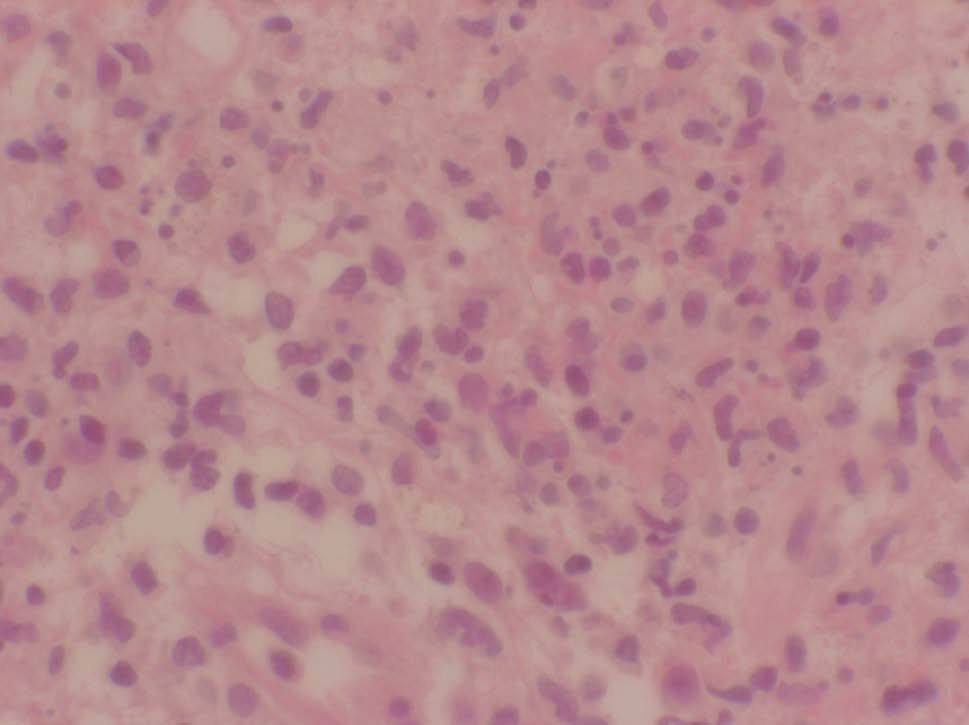
Se realizó una nueva biopsia cutánea de una lesión del tronco que reveló un intenso edema dérmico acompañado de un infiltrado neutrofílico masivo sin presencia de vasculitis (figs. 2 y 3), compatibles con un SS. Se pautó tratamiento esteroideo oral a dosis de 1 mg/kg/día con una rápida resolución de las lesiones cutáneas. Sin embargo, el descenso de la dosis de corticoides conllevó una recidiva de las lesiones cutáneas.
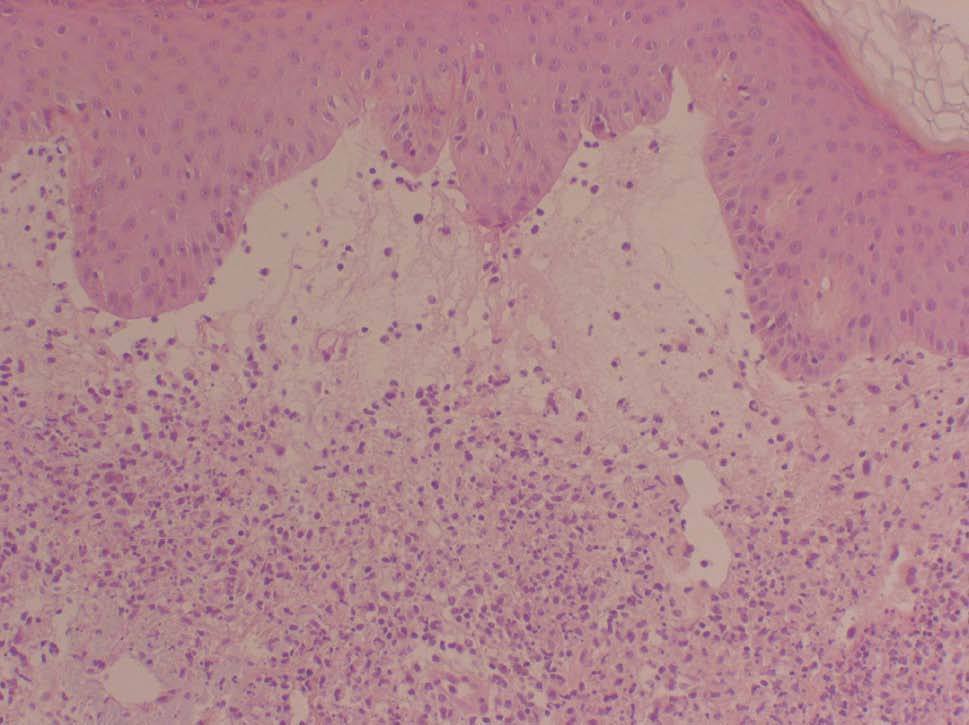
Figura 2. Vista panorámica de la biopsia cutánea que nos muestra el intenso edema de la dermis papilar. (Hematoxilina-eosina, x100.)
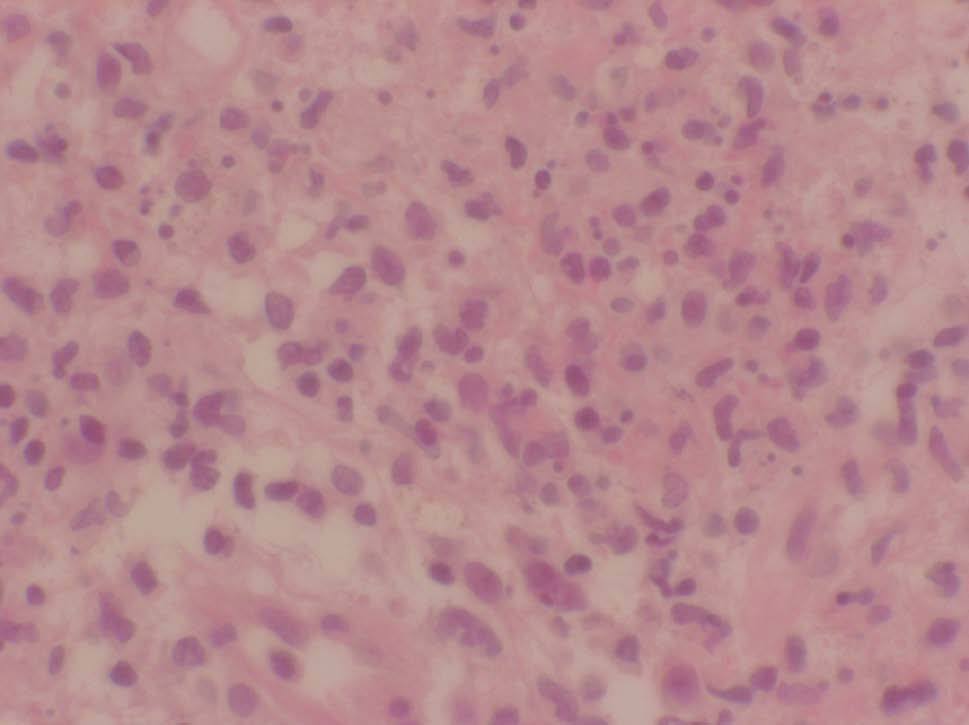
Figura 3. Vista detallada del infiltrado neutrofílico masivo en dermis. (Hematoxilina-eosina, x400.)
En marzo de 1999 presentó un episodio de trombosis venosa profunda y un nuevo episodio de lesiones cutáneas, por lo que fue ingresado. Se trató con acenocumarol y se añadió al tratamiento esteroideo ciclosporina a dosis de 5 mg/kg/día con control de su dermatosis.
En julio de 1999 a pesar de mantener el tratamiento con ciclosporina presentó un nuevo brote de lesiones cutáneas de SS acompañado de aftas orales. Ingresó nuevamente para tratamiento y durante el mismo desarrolló una neumonía nosocomial y shock séptico que requirió ingreso en la unidad de cuidados intensivos y ventilación mecánica. Durante este tiempo el tratamiento inmunosupresor fue suspendido por completo y el paciente presentó mínimas lesiones cutáneas. Fue dado de alta en marzo de 2000 con un extenso estudio analítico, radiológico, marcadores tumorales, estudios serológicos e histopatológicos incluyendo una biopsia de médula ósea que fueron normales o negativos, y no revelaron ningún dato que pudiera explicar el origen de la dermatosis neutrofílica.
En mayo de 2000 presentó un nuevo brote de lesiones cutáneas de gran intensidad y nuevamente se pautó tratamiento esteroideo oral acompañado de azatioprina a dosis de 100 mg/día. Al disminuir la dosis de corticoides se produjo un rebrote inmediato de las lesiones cutáneas. Se buscaron entonces otras alternativas terapéuticas como talidomida, ioduro potásico, colchicina o dapsona sin mejoría de las lesiones cutáneas.
En junio de 2001 y coincidiendo con un nuevo brote de lesiones cutáneas presentó una necrosis avascular de la cabeza del fémur por lo que nuevamente fue ingresado. Una analítica realizada durante este ingreso reveló un factor reumatoide de 84 UI/l, una hipergammaglobulinemia policlonal y un ligero descenso del complemento. Se instauró nuevamente tratamiento con prednisona a dosis de 15 mg/día, azatioprina a dosis de 100 mg/24 h y colchicina 1 mg/8 h con remisión de las lesiones cutáneas.
En agosto de 2001 desarrolló una intensa recurrencia de sus lesiones cutáneas con lesiones pustulosas generalizadas, algunas de las cuales presentaban un aspecto hemorrágico. En este momento se realizó una serología para el virus de la hepatitis C que resultó positiva. Se disminuyeron entonces las dosis de prednisona y azatioprina.
En los meses posteriores el paciente desarrolló una pancitopenia progresiva y una coagulopatía, y falleció en junio de 2002.
Los estudios repetidos realizados a nuestro paciente en los 7 años de su enfermedad no demostraron la presencia de una neoplasia sólida o hematológica, una infección activa u otros hallazgos que pudieran explicar la causa del SS. La cronicidad y evolución del cuadro fue etiquetada como un síndrome de respuesta inflamatoria sistémica crónico idiopático.
Discusión
Nuestro paciente fue diagnosticado de SS por cumplir 2 criterios mayores y 2 menores de los propuestos por Su y Liu, comienzo agudo de placas eritemato-edematosas dolorosas, edema dérmico acompañado de un infiltrado compuesto de forma predominante por neutrófilos sin vasculitis leucocitoclástica, síntomas prodrómicos como fiebre y buena respuesta a tratamiento esteroideo.
La evolución del SS en este caso es atípica por su cronicidad y largo tiempo de evolución, por su gran intensidad y por el desenlace fatal del enfermo. El progresivo deterioro del paciente se debe a que el SS se desarrolla acompañando a un síndrome de respuesta inflamatoria sistémica crónico. La etiología de ambos no se ha podido demostrar en la gran cantidad de estudios realizados en los 7 años de evolución del cuadro clínico. Un estímulo antigénico de naturaleza desconocida probablemente fuera el responsable de la cronicidad del SS y del síndrome de respuesta inflamatoria sistémica crónica.
La positividad serológica para el virus de hepatitis C, demostrada de forma tardía en nuestro paciente, podría explicar el cuadro clínico; sin embargo, existen varios hechos que no nos permiten poder relacionar ambos cuadros:
1.La prevalencia del virus C en la población general es alta y su asociación con SS excepcional19.
2.No se ha descrito el síndrome de respuesta inflamatoria sistémica en relación con el virus C.
3.La positividad antigénica para el mismo no conlleva su participación en el cuadro. Dada la mala evolución del paciente no se realizó un estudio más profundo para determinar las copias de ARN del virus C.
Durante la infección nosocomial que requirió ingreso en la unidad de cuidados intensivos, nuestro paciente no presentó lesiones de SS. Posteriormente, y acompañando a la recuperación inmunitaria del enfermo, aparecieron de nuevo. Este hecho puede ser explicado como un fenómeno similar al síndrome de reconstitución inflamatoria ocurrido en los pacientes infectados por el virus de la inmunodeficiencia humana (VIH) tras el tratamiento con la terapia antirretroviral de alta actividad (TARGA). Ya se ha descrito de forma previa un caso de SS en un paciente VIH tras el tratamiento con la TARGA20-22.
El SS como una dermatosis que compromete la vida ha sido raramente descrito6-13. Si excluimos las asociaciones a enfermedades graves que pueden llevar a la muerte del enfermo, sobre todo las neoplasias hematológicas, existen pocos casos de SS de evolución fatal. Éstos se deben a la afectación de estructuras vitales por la infiltración neutrofílica, como las fibras elásticas de las grandes arterias (síndrome de Marshall), fibras elásticas cardiacas, miocardio, sistema de conducción del corazón o hígado.
Hay muy pocos casos de SS asociado a síndrome de respuesta inflamatoria sistémica16-18, y ninguno de ellos fue mortal. Creemos por tanto que nuestro caso es el primero de SS asociado a un síndrome de respuesta inflamatoria sistémica de evolución fatal.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Jesús del Pozo. Servicio de Dermatología.
Complejo Hospitalario Universitario Juan Canalejo.
Xubias de Arriba, 84.
15006 La Coruña. España.
Correo electrónico: del_pozo@canalejo.org
Aceptado el 26 de octubre de 2006.



