




INTRODUCCION
El pénfigo es una enfermedad ampollosa autoinmune órgano-específica, poco frecuente, que afecta a la piel, mucosas y anejos, caracterizada histopatológicamente por acantolisis, inducida por la presencia de autoanticuerpos patogénicos frente a diversas proteínas desmosómicas. El pénfigo se ha dividido en dos grandes grupos, el pénfigo vulgar (PV) y el pénfigo foliáceo (PF), según la localización de la ampolla. Además, cada uno de ellos tiene su variante clínica de pénfigo vegetante y pénfigo eritematoso, respectivamente. En los últimos años se han descrito nuevas variantes de pénfigo: el pénfigo paraneoplásico (PPN), el pénfigo IgA (inmunoglobulina A) y el pénfigo herpetiforme (PH). El avance de las técnicas de biología molecular ha permitido conocer con precisión los diferentes antígenos frente a los cuales van dirigidos los autoanticuerpos, y poner a punto las técnicas de análisis de inmunoabsorción ligado a enzimas (ELISA) para el diagnóstico.
HISTORIA
Sauvages (1760) introdujo el término pénfigo en su clasificación de enfermedades ampollosas, quizá refiriéndose al eritema exudativo multiforme. Posteriormente, Wichman (1791) fue el primero en describir el pénfigo como una enfermedad crónica ampollosa, pero Willan (1808) lo consideró como una erupción ampollosa de corta duración. Cazenave (1844) describió el PF. Sin embargo, fue Hebra (1869) el que ordena diversas entidades considerando el pénfigo como una enfermedad crónica ampollosa, desechando de tal denominación las formas agudas de la enfermedad, dividiendo al pénfigo en vulgar y foliáceo, similar al descrito por Cazenave. El pénfigo vegetante fue descrito por Isidor Neumann (1886). Al comienzo de la siguiente década Besnier (1891) y más tarde Brocq (1902) tratan de poner más orden en el entonces complejo capítulo de los pénfigos y realizan dos aportaciones fundamentales: la histopatología del pénfigo y el signo de Nikolsky. Auspitz (1881) reconoce por primera vez que el pénfigo está caracterizado histológicamente por la desaparición de los llamados puentes intercelulares entre los queratinocitos, acuñando el término acantolisis para describir este fenómeno. No obstante, este hallazgo fue ignorado durante mucho tiempo hasta que Jean Darier y Achille Civatte (1943) demostraron su importancia diagnóstica. Los hallazgos histopatológicos de Lever (1953), que introducen el término penfigoide como una enfermedad ampollosa sin acantolisis, inician la era moderna de las enfermedades ampollosas 1,2.
El PH fue descrito por Jablonska3 en 1975 como dermatitis herpetiforme con acantolisis, pénfigo con repuesta a las sulfamidas o enfermedad mixta ampollosa. Desde entonces se han publicado numerosos casos, y es una entidad aceptada por todos los autores, aun cuando su posición nosológica con relación al pénfigo sigue siendo controvertida 4. Algunos investigadores lo consideran diferente del pénfigo clásico basándose en sus peculiaridades clínicas y en su pronóstico benigno, pero otros los describen como variantes del PF o del PV.
El primer caso de pénfigo IgA fue descrito en 1982 con el nombre de dermatosis pustulosa subcórnea e IgA monoclonal 5, aunque ya se había encontrado un depósito de IgA similar en la descripción de Sneedon y Wilkinson en 1979. Desde entonces se han descrito numerosos casos bajo diferentes nombres como dermatosis IgA neutrofílica intraepidérmica, dermatosis IgA intercelular, PF IgA, PH IgA y pustulosis intraepidérmica IgA, entre otros 6.
El PPN fue descrito como tal en 1990 7, aunque no es una enfermedad nueva, y casos semejantes habían sido reflejados previamente en la literatura médica bajo designaciones diversas como casos atípicos de PV, raros casos de eritema multiforme con anticuerpos tipo pénfigo, o enfermedad paraneoplásica ampollosa 8.
EPIDEMIOLOGIA
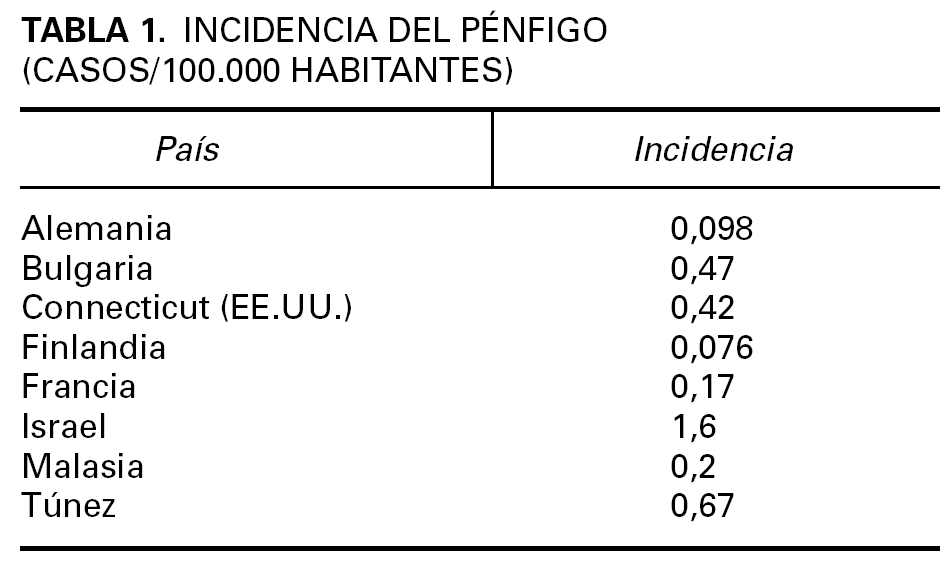
Los datos epidemiológicos del pénfigo son limitados 9-15. Es una enfermedad infrecuente cuya incidencia, prevalencia y caracteres epidemiológicos son variables en función de factores geográficos y étnicos (tabla 1).
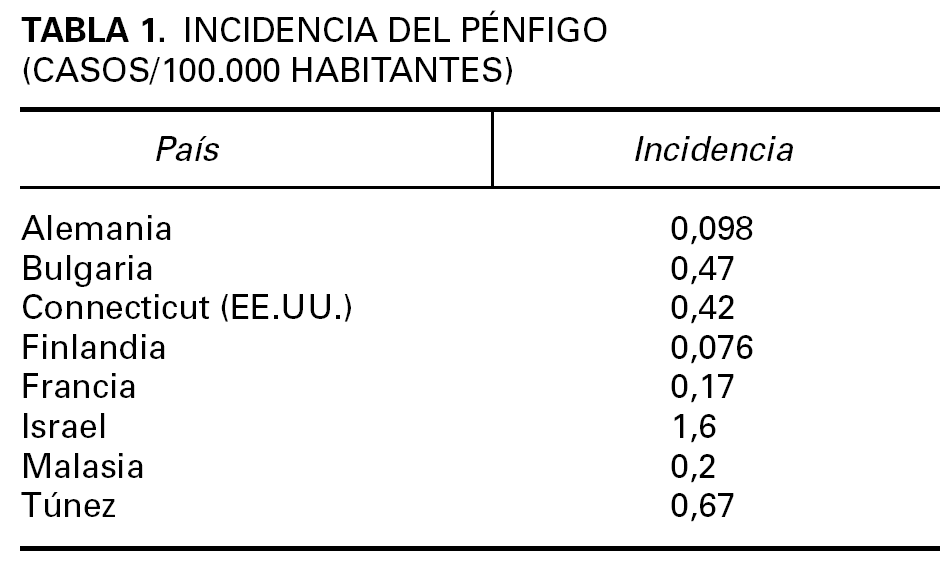
La incidencia del PV se ha estimado entre 0,1 y 0,5 por 100.000 personas y año. Afecta por igual a ambos sexos, con un pico de incidencia entre la cuarta y la sexta década. Es más frecuentes en determinadas razas, singularmente la raza judía de ascendencia ashkenazi en el área del Mediterráneo, donde la incidencia oscila entre 1,6 y 3,2 por 100.000 personas y año. En Israel, Estados Unidos y países de Europa predomina el PV sobre el PF, pero en algunos países del Norte de Europa la frecuencia del PF es algo mayor.
La incidencia del PF no endémico es de 0,5 por 100.000 personas y año en Francia, y afecta por igual a ambos sexos, habitualmente en la edad media de la vida 16. La forma endémica del PF, también conocida como «fogo selvagem», se ha descrito en países en desarrollo, en particular Sudamérica, India, África 17,18, y en la península arábiga. Los datos epidemiológicos, la distribución por edades, los casos familiares y la frecuencia de ciertos haplotipos de antígenos de histocompatibilidad (HLA) muestran características peculiares del fogo selvagem que los distingue del PF no endémico. En Brasil el fogo selvagem afecta a niños y adultos jóvenes de ambos sexos con un pico de incidencia en la segunda y tercera década, con muchos casos familiares. La mayoría de los pacientes se encuentran en áreas rurales, que viven en casas rústicas y expuestos a insectos hematófagos 19. Recientemente se ha identificado un nuevo foco de PF endémico en Brasil en la región de Limão Verde donde la prevalencia de la enfermedad es de un 3,4 % 20. Se ha descrito un cuadro clínico semejante en Colombia 21, que afecta a varones entre 40 y 60 años y a mujeres posmenopáusicas. En Túnez las series de casos 17,22, indican que es más frecuente en mujeres jóvenes, en áreas rurales, sin predominio familiar, habiéndose puesto en relación con la utilización de cosméticos tradicionales 23.
CLASIFICACION
Clásicamente la clasificación se ha realizado atendiendo a criterios clínicos e histopatológicos, que muestra una concordancia marcada con el antígeno responsable, como se puede observar en la tabla 2 24. Existen diversas variedades bien definidas clínica e histopatológicamente de pénfigo que reconocen el mismo sustrato antigénico, a pesar de mostrar diferentes manifestaciones clinicopatológicas. Por ejemplo, en el PH los antígenos aislados son los mismos que en el PV cutaneomucoso, a pesar de que las manifestaciones clinicopatológicas son diferentes. Quizás este hecho pudiera ser debido a una expresión diferente de citocinas liberadas por la reacción inmunológica frente a distintos epítopos de las desmogleínas.

ETIOLOGIA
La sintomatología y los caracteres demográficos observados en el pénfigo en diversos países han implicado a factores genéticos y ambientales que contribuyen a la etiopatogenia del pénfigo. El comienzo y desarrollo del pénfigo depende de la interacción entre los factores genéticos predisponentes y los factores inductores del mismo, que condicionan una respuesta inmunológica del individuo contra los desmosomas epidérmicos. A continuación se exponen los factores genéticos y desencadenantes conocidos en la etiopatogenia del pénfigo.
Factores genéticos
En la literatura especializada se han publicado casos de pénfigo familiar, casos de pénfigo en gemelos monozigotos y la presencia de anticuerpos contra la superficie de queratinocitos en el suero de los familiares de los pacientes 25. Sin embargo, también se ha publicado la existencia de pénfigo en únicamente uno de los gemelos monozigotos, observación que junto a la escasa frecuencia del pénfigo familiar sugiere que la predisposición genética no es suficiente para la aparición de la enfermedad, y son también importantes los factores externos.
Se ha observado una asociación entre los antígenos de los complejos mayores de histocompatibilidad y el pénfigo. Se piensa que los genes del HLA de clase II tienen un papel importante en la inmunopatogenia del pénfigo. Se ha demostrado que en el PV predominan los alelos HLA-DR4 (DRB1*0402, DRB1*0406), HLA-DR14 (DRB1*1401, DRB1*1405) y HLA-DQ1 (DQB1*0503) en grupos étnicos diferentes. En los judíos ashkenazi con pénfigo predomina el antígeno HLA-DR4, de los cuales el 95 % portan el alelo DRB1*0402. El polimorfismo alélico en el PV se traduce en alteraciones menores en las secuencias de aminoácidos de la cadena β1 del HLA-DR que aumenta la capacidad de presentar los autoantígenos, a diferencia de lo que ocurre en individuos sin PV 26-28. Se encontró una fuerte asociación entre el PF no endémico y los alelos del complejo mayor de histocompatibilidad (MHC) de clase II, sobre todo con los alelos DRB1*0102 y DRB1*0404. Estos resultados están en concordancia con aquéllos observados en el PF endémico, donde se ha observado una asociación con DRB1*0102, DRB1*0404, DRB1*1402 y DRB1*140629.
Sólo se han descrito referencias aisladas al papel de los alelos del MHC de clase I 30, aunque últimamente se pretende darles mayor importancia 31. Los estudios iniciales en el PV mostraron un aumento en la frecuencia del HLA-A10 (A26), y Bw38, pero posteriormente se han observado diferentes antígenos HLA de clase I en función de la raza. En experimentos recientes se ha demostrado una relación entre el HLA-G y el PV 32. Hasta la actualidad no se ha descrito ningún alelo HLA de clase I asociado a PF.
Factores desencadenantes
Fármacos33
El pénfigo inducido por fármacos es infrecuente y son diversos los fármacos que se han implicado como se observa en la tabla 3. La mayor parte de los medicamentos capaces de inducir pénfigo suelen tener un grupo tiol (SH) en su molécula o contienen un enlace disulfuro que potencialmente es capaz de liberar grupos tiol. Otros fármacos tienen en su composición azufre en cuyo metabolismo se pueden también liberar grupos tiol, incluyendo penicilinas, cefalosporinas y piroxicam.

El tiempo transcurrido entre la administración del medicamento y la aparición de la enfermedad puede ser desde pocas semanas hasta varios meses, siendo el periodo mayor para los medicamentos del grupo tiol. Clásicamente se ha descrito una sintomatología semejante al PF o eritematoso, relacionada con medicamentos del grupo tiol. Los casos de PV son cada vez más frecuentes y se asocian con fármacos del grupo no-tiol. Algunas diferencias ayudan a diferenciarlo del pénfigo idiopático. El pénfigo inducido por fármacos generalmente se acompaña inicialmente de lesiones prodrómicas no ampollosas e inespecíficas, como exantemas o eritemas anulares, entre otros, que recuerdan una reacción medicamentosa. Histopatológicamente incluye espongiosis eosinofílica, necrosis epitelial con infiltrado dérmico denso y no es infrecuente que en un mismo enfermo se encuentren ampollas subcórneas y suprabasales en lesiones diferentes e incluso en la misma lesión. Los anticuerpos contra los queratinocitos en piel, mucosa y suero no siempre están presentes y pueden tener anticuerpos frente a otras estructuras no epiteliales. La mayoría de los casos mejoran al suspender la medicación responsable, pero aproximadamente una tercera parte de los inducidos por d-penicilamina necesitan tratamiento esteroideo para controlar la enfermedad.
No se conoce con exactitud el mecanismo por el cual se induce el pénfigo, pero algunos fármacos inducirían una verdadera acantolisis bioquímica, mientras que otros desencadenarían una acantolisis inmunológica. La acantolisis bioquímica se ha relacionado con fármacos del grupo tiol, que produciría una alteración bioquímica directa de la superficie celular del queratinocito que impide la adhesión celular. En la acantolisis inmunológica el medicamento formaría un neoantígeno con la subsiguiente aparición de anticuerpos. Ambos mecanismos no tienen por qué ser mutuamente excluyentes.
Factores hormonales
Se ha especulado con la posibilidad de que factores hormonales modulen la aparición o el curso del pénfigo, basados en algunos casos que han sufrido exacerbación del mismo en el embarazo 34 o su aparición en el mismo 35.
Radiaciones ultravioleta36
El efecto nocivo de la exposición al sol en el comienzo, curso y evolución del pénfigo se ha observado hace ya mucho tiempo. El nombre de fogo selvagem se introdujo para designar la sensación de quemazón de la piel en estos enfermos cuando se exponían al sol. Tanto el PF 37 como el PV 38 se han exacerbado después de la terapia con luz ultravioleta y psoralenos (PUVA) 39, por radiación ultravioleta B (UVB), después de la exposición solar 40, y tras la irradiación con luz ultravioleta 41. Se ha demostrado que la exposición a la luz UVB de la piel sana en el fogo selvagem y PV promueve la acantolisis con el depósito de IgG y C3 en los espacios intercelulares y, como consecuencia de ello, se ha propugnado la abstención de la exposición solar prolongada y la utilización de filtros solares como parte del tratamiento.
Algunos autores 42 han revisado los tres posibles mecanismos propuestos para la inducción de acantolisis en las lesiones de pénfigo inducidas por el sol, que no tienen por qué ser mutuamente excluyentes. La exposición solar al dañar la membrana celular puede conducir a una mayor capacidad de unión de los antisueros a las superficies del queratinocito, de manera semejante a lo que ocurre en el lupus eritematoso. El segundo mecanismo propone que la luz UV puede modular seniales intracelulares una vez que los anticuerpos patógenos se liguen a la desmogleína de los queratinocitos a través de la elaboración de factores todavía no descritos. Una tercera hipótesis considera que los mediadores inflamatorios liberados de los queratinocitos y/o mastocitos por la exposición a luz UVB, entre los que se encuentra interleucina 1 (IL-1), IL-8, el factor de necrosis tumoral alfa (TNF-α) y el factor estimulante de colonias macrocíticas y granulocíticas (GM-CSF), podrían ser los responsables de la aparición de las lesiones ampollosas.
Dermatitis de contacto 43
Existen algunas publicaciones de casos de pénfigo que se desarrollan después de una dermatitis de contacto a diversos productos. Inicialmente fue descrita en relación con el contacto con el ajo, también se ha puesto en relación con otras sustancias como determinados pesticidas, fenoles y, recientemente, tiuranes e imiquimod 44,45. El mecanismo patogénico propuesto es una acción directa de la sustancia desencadenante 46, que es capaz de producir una alteración en la superficie cutánea, con la formación de neoantígenos.
Radiaciones ionizantes 47-49
En la literatura médica se han descrito algunos casos de pénfigo inducidos por radioterapia. La mayoría de ellos presenta un cáncer subyacente planteando el problema del papel que podría desempeñar la neoplasia en el desarrollo de la enfermedad. Sin embargo, en algún caso se utilizó la radioterapia para tratar un epitelioma basocelular, lo cual hace poco probable que la neoplasia tenga algo que ver con el pénfigo.
Cicatrices quirúrgicas 50-52
Se han descrito algunos pénfigos secundarios a injertos, cirugía mamaria o rinoplastia. El intervalo entre la intervención quirúrgica es muy variable, entre 2 meses y 3 años, aun cuando existe un caso con la aparición de pénfigo 40 años después. Se ha sugerido que pudiera ser una forma de fenómeno isomórfico, que alteraría la capacidad antigénica de las estructuras cutáneas.
Quemaduras
Las quemaduras se han implicado también como factor desencadenante en algunos artículos publicados 53 tras la aparición transitoria de autoanticuerpos semejantes a los del pénfigo en el suero de algunos pacientes quemados.
Dieta 54
Algunos autores han sugerido que los alimentos con una estructura molecular semejante al grupo tiol de los medicamentos como los derivados del ajo, cebolla o los puerros pueden desencadenar el pénfigo. Los alimentos con grupos isotiocianatos como la mostaza, los fenoles como el aspartamo usado como edulcorante natural o los taninos como la mandioca y el mango, se han implicado en el pénfigo. Se ha sugerido que la eliminación o reducción de estos alimentos pueden disminuir la morbilidad de la enfermedad, aunque se desconoce el mecanismo por el que actúan.
Infecciones 55
Numerosos estudios han tratado de demostrar la relación entre las diversas infecciones y el pénfigo. Los virus han sido los agentes más implicados y con las técnicas de reacción en cadena de la polimerasa (PCR) se ha sugerido que el virus del herpes simple, el virus de Epstein-Barr, el citomegalovirus y últimamente el virus herpes humano tipo 8 pueden ser capaces de inducir o exacerbar el pénfigo en sujetos susceptibles 56. A causa de las falsas positividades sin correlación clínica de la PCR, estos resultados deben ser interpretados con mucha cautela. Los virus podrían ser infecciones oportunistas en pacientes inmunosuprimidos, pero en algunos casos podrían tener una relevancia patogénica. Las infecciones por estafilococos, Proteus vulgaris y Pseudomonas en relación con el PV parecen más bien una complicación del mismo. Recientemente se conoce que las toxinas exfoliativas del estafilococo dorado actúan sobre la desmogleína 1, que es la responsable de la ampolla en el pénfigo 57.
PATOGENIA
En las últimas décadas numerosos e importantes avances han permitido un conocimiento más detallado de la fisiopatología de los diferentes tipos de pénfigo 58-61. Los hallazgos se agruparán en los apartados de antígenos, anticuerpos, inmunidad celular y mecanismos fisiopatológicos propuestos para la aparición de acantolisis.
Antígenos del pénfigo
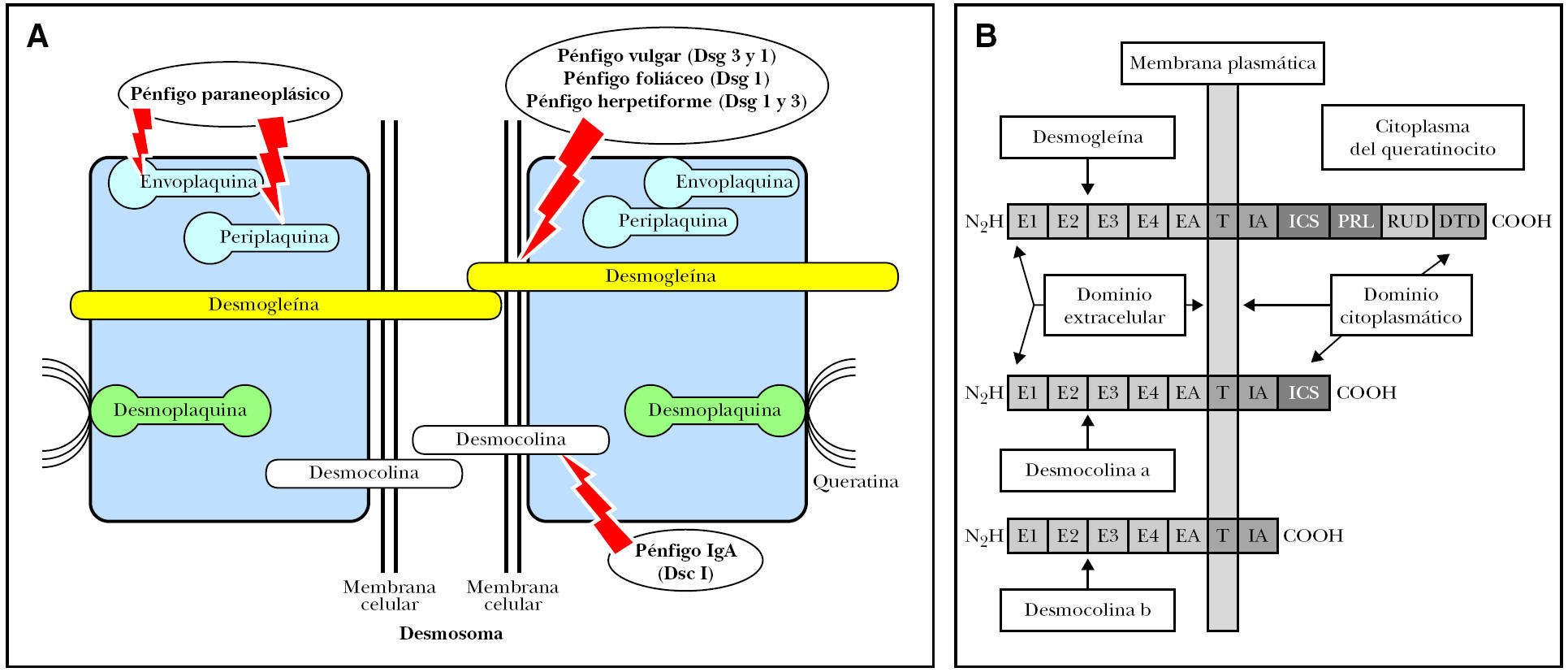
La estructura epidérmica primariamente implicada en el pénfigo es el desmosoma 62, una de cuyas funciones es el mantenimiento de las uniones intercelulares intraepidérmicas. Los desmosomas son estructuras muy organizadas con tres tipos de moléculas: cadherinas, proteínas de armadillo y plaquinas. Dentro de las cadherinas 63,64 se encuentran dos grupos distintos de proteínas transmembrana denominadas desmocolinas y desmogleínas, cada una de ellas constituidas por tres isoformas (1 a 3). Recientemente se ha descrito una cuarta isoforma 65. La placoglobina y la placofilina son proteínas intracitoplásmicas derivadas de la familia de los supergenes del armadillo, que conectan con la desmogleína y desmocolina, regulando la actividad adhesiva de estas moléculas. La familia de las plaquinas son proteínas intracelulares que están formadas por la desmoplaquina, envoplaquina, periplaquina y plectina. La desmoplaquina es la más abundante y parece que es la proteína que liga directamente los filamentos intermedios de queratina (figs. 1A y B).
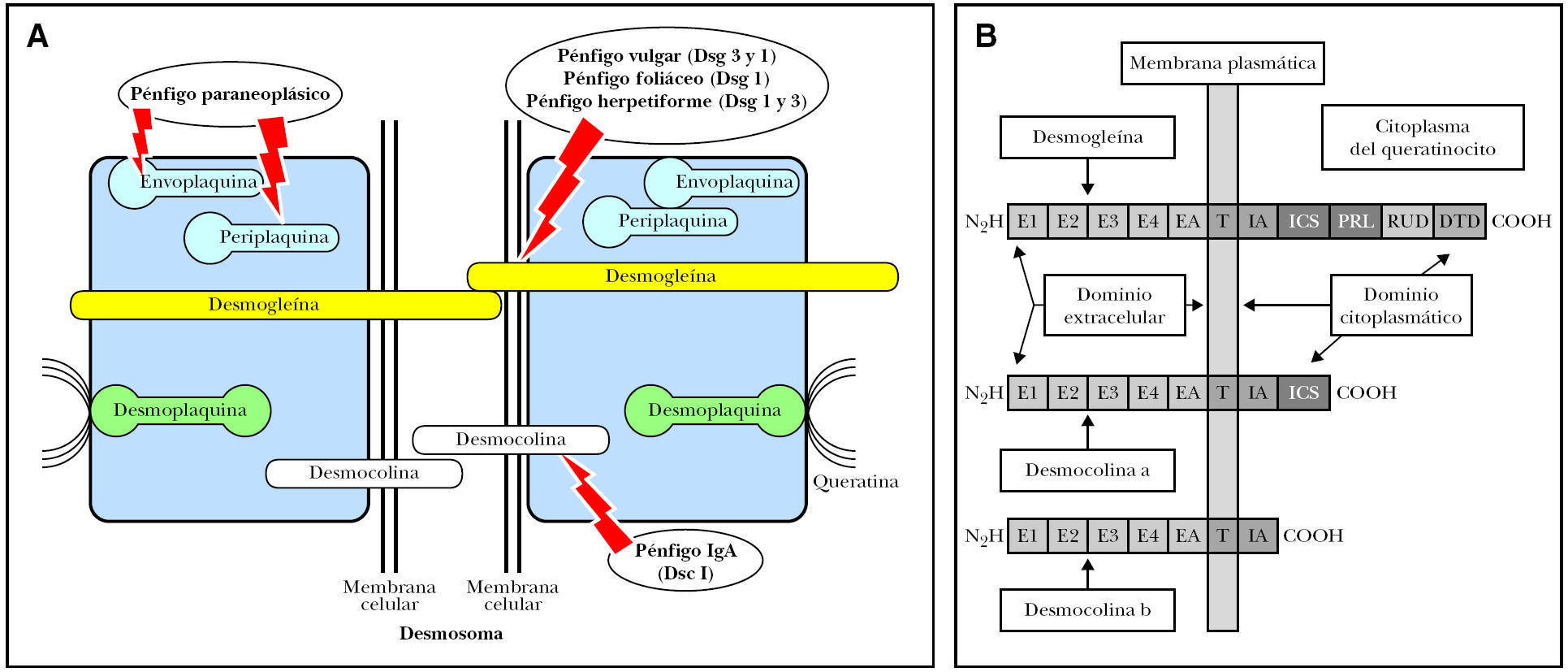
Fig. 1.--Esquema de la desmogleína y la desmocolina en el queratinocito. E1-E4: elementos repetidos extracelulares; EA: dominio de anclaje extracelular; IA: dominio de anclaje intracelular; ICS: segmento de unión de la placoglobina; PRL: segmento rico en prolina; RUD: dominio de unión repetido; DTD: dominio terminal de la desmogleína.
En 1987, el grupo de Stanley demostró que los autoanticuerpos en el PF reconocen a la desmogleína 1 de 160 kDa 66, y en el PV a la desmogleína 3 de 130 kDa 67. El PV con predominio de lesiones mucosas tiene anticuerpos contra la desmogleína 3 y cuando aparecen lesiones cutáneas, presenta también anticuerpos contra la desmogleína 1.
Posteriormente, mediante inmunoprecipitación se aislaron diversas proteínas en la epidermis humana de diferentes pesos moleculares utilizando anticuerpos de sueros de pacientes con PV y PF 68. Los epítopos autoinmunes, tanto en el PV como el PF se encuentran en el extremo aminoterminal de las desmogleínas 1 y 3 69,70, en el primer y segundo dominio extracelular. Aunque otras moléculas de la superficie de los queratinocitos, como los receptores de la acetilcolina, pueden modular la adhesión celular, su implicación directa en la fisiopatología del pénfigo es controvertida 71,72.
Los estudios sobre la distribución en la piel normal de la desmogleína 1 y 3 han demostrado que la desmogleína 1 se expresa en la totalidad del espesor de la epidermis, siendo mínima su expresión en mucosas. La desmogleína 3 se expresa únicamente en las capas más profundas de la epidermis, mientras que en la mucosa se expresa en todo su espesor. Se ha propuesto la hipótesis compensatoria para explicar los hallazgos clínicos: en el PV mucoso los anticuerpos contra la desmogleína 3 condicionarían lesiones en mucosas debido a la ausencia de la compensación de la desmogleína 1 en la mucosas, y no presentarían lesiones en la piel por la compensación de la desmogleína 1. La presencia de anticuerpos contra la desmogleína 1 en PV hace que la compensación no sea posible, con aparición de lesiones cutáneas. En el PF los anticuerpos contra la desmogleína 1 no causan lesiones mucosas debido al efecto compensador de la desmogleína 3, pero al encontrarse sólo en la capa basal epidérmica no podría evitar la aparición de lesiones cutáneas 73,74.
En 1997 se desarrolló un ratón genéticamente modificado que no sintetizaba desmogleína 3, con la piel y mucosas normales en el nacimiento, pero en 15 o 20 días se producían erosiones con acantolisis de la mucosa oral y costras con acantolisis en la piel traumatizada 75. Posteriormente se demostró que las proteínas recombinantes desmogleína 1 y 3 con su configuración original generadas en un sistema de expresión por baculovirus, eran capaces de adsorber todos los anticuerpos patógenos del suero del PV y PF 76,77. Por lo tanto, los autoanticuerpos contra la desmogleína 1 y 3 tienen valor patogénico. Estas proteínas recombinantes han permitido poner a punto la técnica de ELISA para detectar autoanticuerpos circulantes en el suero del pénfigo. La técnica de ELISA ha demostrado ser muy sensible y específica, y ha contribuido al progreso en el diagnóstico, clasificación y fisiopatología del pénfigo 78.
En el año 2000 investigadores japoneses del grupo de Amagay desarrollaron el primer modelo animal activo de la enfermedad en un ratón genéticamente deficiente en desmogleína 3, que era inmunizado con desmogleína 3 recombinante para producir IgG contra ella. Los esplenocitos del ratón sensibilizado eran transferidos a un ratón inmunodeficiente, y en él tenía lugar la producción de antoanticuerpos por la transferencia de las células inmunes con lesiones clínicas e histológicas de PV 79.
Anticuerpos del pénfigo
Los enfermos con PV poseen autoanticuerpos en la piel lesional y en el suero frente a estructuras epidérmicas. Utilizando técnicas de inmunofluorescencia indirecta, Beutner y Jordan demostraron autoanticuerpos circulantes frente a antígenos intercelulares de la piel en 1964 80. Posteriormente demostraron 81 que estos autoanticuerpos se unían a los espacios intercelulares de la epidermis utilizando la técnica de inmunofluorescencia directa. Más tarde se demostró que los anticuerpos tienen valor patogénico en todas las formas de pénfigo, al observarse una correlación entre el título de autoanticuerpos y la actividad de la enfermedad 82. La plasmaféresis, al eliminar los autoanticuerpos circulantes consigue remisiones a corto plazo 83. Los neonatos con PV nacidos de madres con PV muestran remisión espontánea de las ampollas y desaparición de los autoanticuerpos semanas después del parto 84,85. Los estudios experimentales in vitro han demostrado que el suero de enfermos con pénfigo era capaz de inducir acantolisis 86 en la piel humana cultivada. El autoanticuerpo responsable era de clase IgG 87. Estos autoanticuerpos, mediante transferencia pasiva, son capaces de inducir acantolisis con ampollas cuando son inyectados intraperitonealmente a ratones neonatales reproduciendo clínica, histopatológica, ultraestructural e inmunológicamente una enfermedad semejante a la de los seres humanos, demostrándose así el papel patogénico in vivo 88.
Los anticuerpos policlonales del PV son principalmente del tipo IgG4 aunque pueden observarse otros subtipos de inmunoglobulinas, en contraste con los familiares en los que apenas se detecta IgG489. En el PV mucoso los anticuerpos IgG1 e IgG4 reaccionan con un epítopo conformacional de la desmogleína 3. En el PV cutáneo y mucoso aproximadamente el 50 % de los enfermos se observan anticuerpos IgG contra la desmogleína 1, además de los anticuerpos antidesmogleína 3. En el PV generalizado los anticuerpos IgG1 parecen reconocer un epítopo lineal en el ectodominio de la desmogleína 3. En contraste, la IgG4 reconoce tanto epítopos conformacionales como lineales en la desmogleína 3 90. Además de los autoanticuerpos IgG, los autoanticuerpos IgA y ocasionalmente los IgE reactivos frente a la desmogleína 3 están presentes en las fases aguda y crónica del PV 91.
En el PF el análisis de anticuerpos revela que los autoanticuerpos IgG4 e IgG1 son los isotipos más frecuentes frente a la antidesmogleína 1 92, 93 mientras que sólo el 7 % de los pacientes muestra actividad frente a la desmogleína 3 94. Los epítopos reconocidos por los anticuerpos del PF son conformacionalmente sensibles 95 y dependientes del calcio 66. En el PF los autoanticuerpos IgG4 reaccionan con un epítopo conformacional mientras los anticuerpos IgG1 reaccionan con un epítopo lineal del ectodominio de la desmogleína 1 90. El valor de IgG4 está estrechamente relacionado con la actividad de la enfermedad, mientras que la concentración de IgG1 se mantiene constante a lo largo de la misma. En la zona de Limão Verde (Brasil) se han detectado anticuerpos IgG antidesmogleína 1 en el 97 % de los enfermos, pero también en el 55 % de los sujetos normales de zonas endémicas, porcentaje que progresivamente disminuye en las áreas más periféricas. En los sujetos normales se produce una respuesta IgG1 e IgG4, mientras que los pacientes tienen niveles similares de IgG1 pero mucho más aumentados de IgG4. Los pacientes en remisión presentan una débil respuesta IgG4 y la enfermedad activa cursa con concentraciones muy aumentadas 20. En un subgrupo de pacientes con PF sólo se detectaron autoanticuerpos IgG1 que reaccionaron con epítopos lineales antidesmogleína 1 en todo el curso de su enfermedad, sin que se observasen hallazgos clínicos diferenciales con respecto a los que tienen autoanticuerpos IgG4 96. Se han detectado autoanticuerpos contra el extremo carboxiterminal del dominio denominado «EC5» de la desmogleína 1 en individuos sin enfermedad cutánea. Esta respuesta autoinmune en individuos genéticamente predispuestos puede dirigirse al extremo aminoterminal de los dominios EC1 y EC2 de la misma molécula, condicionando el inicio de la enfermedad. Los niveles de los autoanticuerpos contra EC1 y EC2 se correlacionan bien con la actividad clínica de la enfermedad 97.
Inmunidad celular
Los linfocitos T tienen un papel importante en la inducción y regulación de los niveles plasmáticos de anticuerpos en el pénfigo 98. Los estudios realizados in vitro y en animales han demostrado la necesaria colaboración entre los linfocitos T CD4 estimulados por la desmogleína 3 y las células B para inducir la producción de anticuerpos 99. En pacientes con PV se han detectado tanto linfocitos Th1 como Th2 específicos contra la desmogleína 3, con niveles constantes aumentados de Th2 en las distintas fases y predominio de los Th1 en la fase activa crónica. La proporción de células Th1/Th2 condiciona una respuesta inmunológica policlonal de anticuerpos IgG1 e IgG4 frente a tres fragmentos antigénicos del ectodominio de la desmogleína 3, posiblemente un péptido compuesto por 15 aminoácidos 100. Las células CD4 α/β segregan citocinas con un patrón del tipo Th2 al contactar con la desmogleína 3 con restricción para los alelos del HLA DRB1*0402 o DRB1*1401 101 activando a los linfocitos B para sintetizar IgG4. Se han detectado niveles aumentados de IL-10, IL-6 y TNF-α en el suero de pacientes con PV en comparación con los sueros de controles normales 102,103. Los portadores sanos de los alelos HLA-II DRB1*0402 y DQB1*0503 asociados al PV presentan linfocitos Th1 que son estimulados por la desmogleína 3, lo que condiciona la aparición de autoanticuerpos IgG1, IgG2 e IgG3. Los linfocitos CD8 también responden a la desmogleína 3 del PV secretando in vitro IL-2 e interferón γ (IFN-γ), pero su función es desconocida 104.
Mecanismos implicados en la acantolisis
Se están investigando los acontecimientos que tienen lugar en el pénfigo después de la unión del anticuerpo con la desmogleína 1 y 3 para provocar acantolisis. Se especula que la rotura final de las uniones intercelulares pueda ser debida a la acción directa de los anticuerpos sobre las desmogleínas del queratinocito que puede condicionar el deterioro estérico de la desmogleína 3 y, como consecuencia, interferir en la función de adhesión intercelular o con su papel de ensamblaje de los desmosomas. La acantolisis se puede producir también de forma indirecta por diversos mecanismos como la activación de las señales transmembranosas tras la fosforilización de esas proteínas, una alteración del balance entre la desmogleína 1 y 3 en los desmosomas epidérmicos, la activación del complemento y el sistema plasminógeno-plasmina.
El inicio de la acantolisis puede estar condicionado directamente por la unión de los anticuerpos del PV al ectodominio de la desmogleína 3, con agrupamiento posterior e internalización del complejo IgG-antígeno 105. La unión de los anticuerpos a epítopos específicos de la desmogleína 3 condicionaría el deterioro estérico de la desmogleína 3 con pérdida de su función de adhesión intercelular 106. El sitio de unión de los anticuerpos patogénicos es el dominio extracelular aminoterminal de la desmogleína 107. Recientemente en ratones se ha observado que la unión de la IgG a la desmogleína 3 produce directamente una hendidura en la superficie apical del queratinocito basal sin retracción de la queratina 108.
En los estudios realizados, ni el complemento 109,110 ni las enzimas proteolíticas 111 son esenciales para el desarrollo de la acantolisis intraepidérmica, aunque no puede descartarse su participación en la formación de ampollas. En un sistema de cultivo celular se ha observado que la IgG del PV condiciona la activación del inositol trifosfato intracelular, aumento del calcio intracelular, el incremento de la actividad de la proteína C cinasa y la fosforilación de la desmogleína 3 112. La inhibición de las proteínas involucradas en la transmisión de señales intracelulares tras la unión de IgG a la desmogleína previene la acantolisis en un modelo murino 113. La placoglobina, una proteína de la placa del desmosoma, tiene un papel importante en la acantolisis inducida por IgG en el PV 114.
MANIFESTACIONES CLINICAS
Pénfigo vulgar
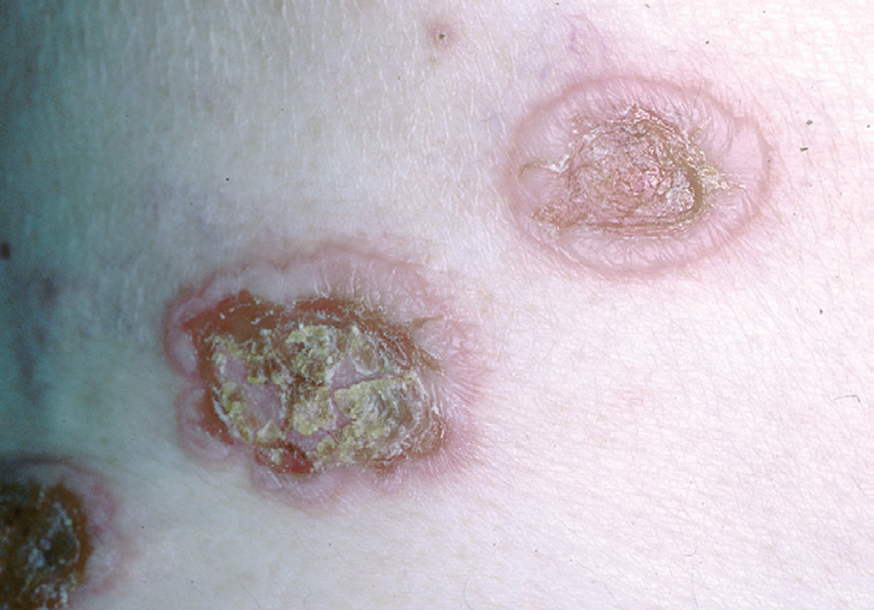
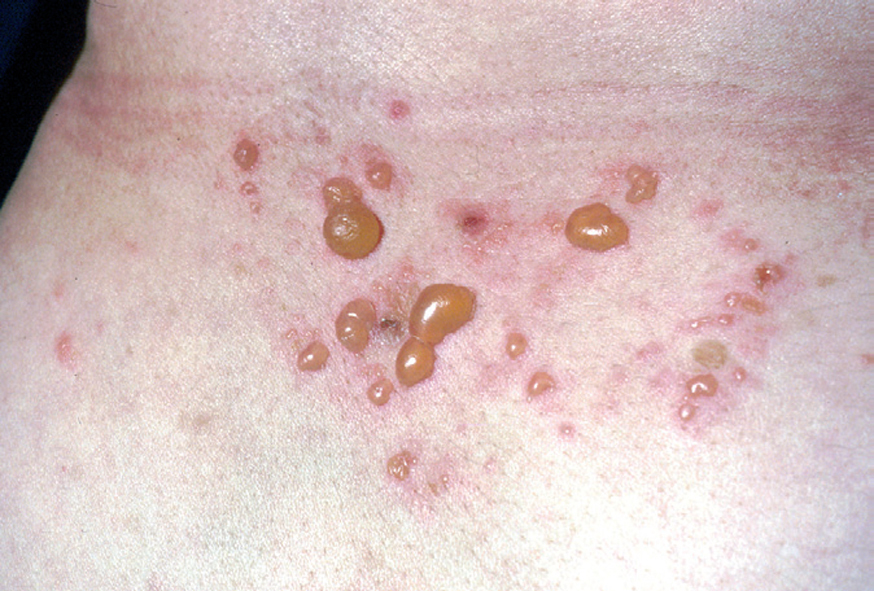
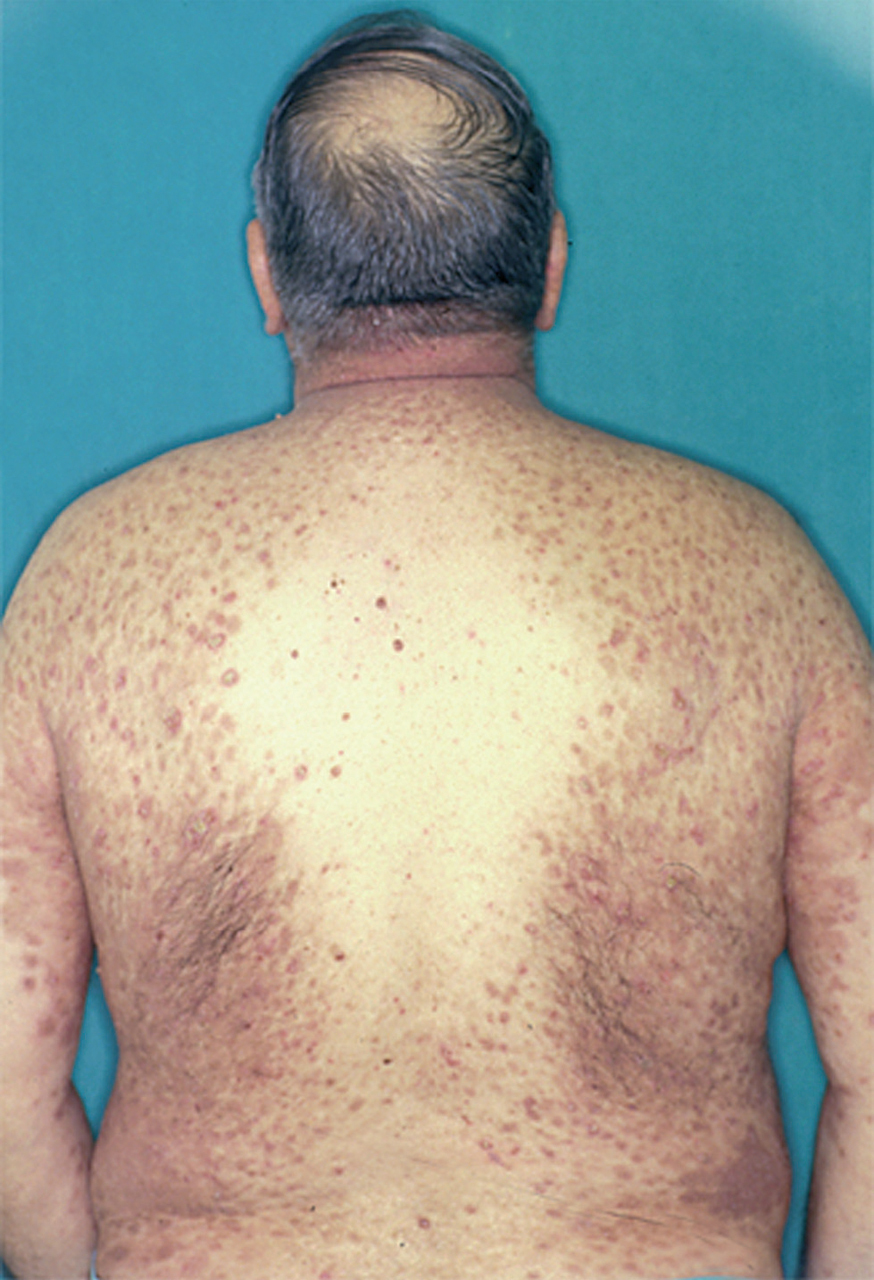
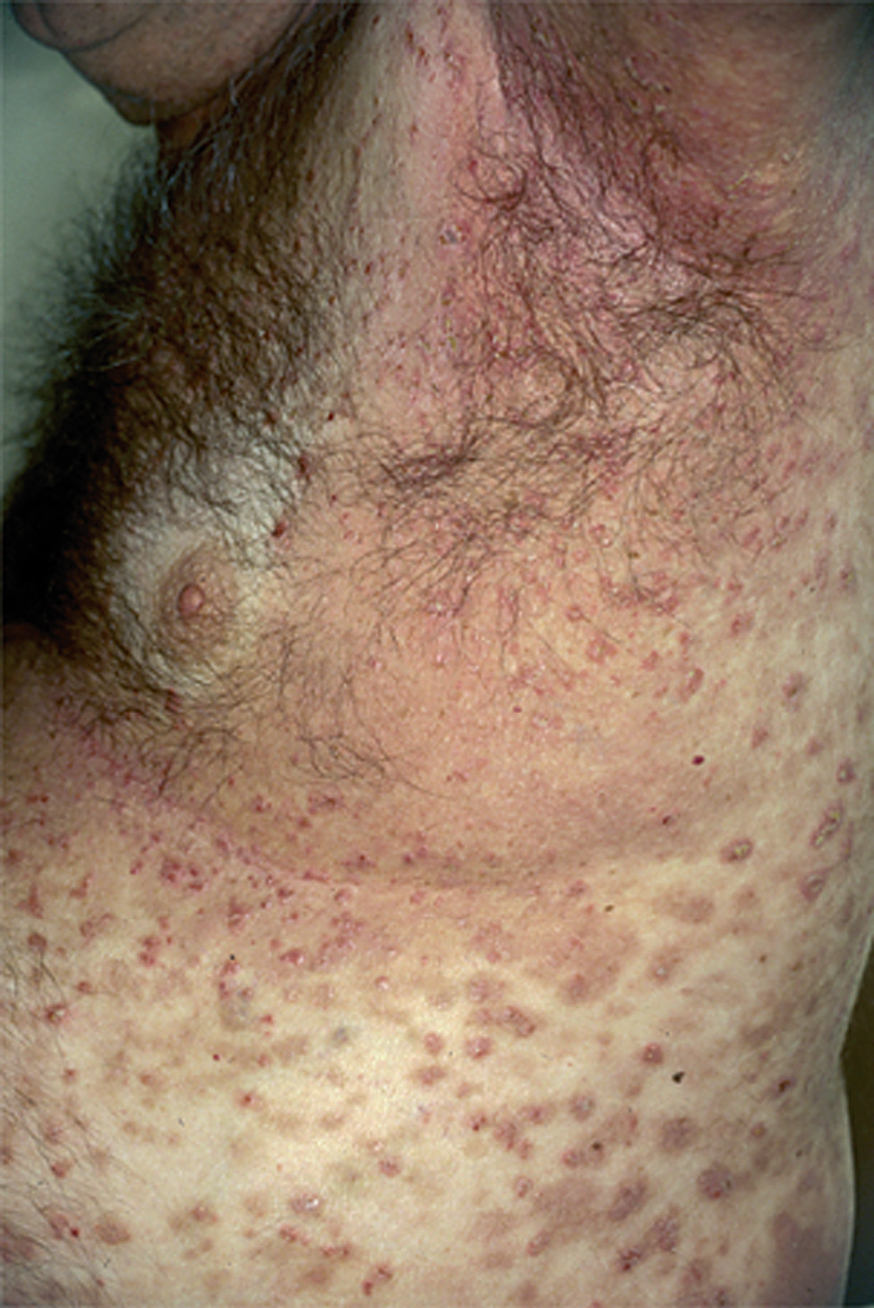
La ampolla es el elemento eruptivo primordial y habitualmente exclusivo 115-118. Tiene un tamaño variable entre el de un guisante y el de una nuez, de color transparente, amarillento o hemorrágico (fig. 2). En ocasiones el contenido de la misma puede ser seroso y purulento. La ampolla habitualmente es de consistencia flácida (fig. 3), aunque en ocasiones se encuentra a tensión. Estas lesiones pueden confluir con ampollas vecinas para adquirir mayor tamaño, o pueden aumentar periféricamente de tamaño dejando en el centro una placa erosivo-costrosa (fig. 4). Clásicamente se ha establecido como característico el hecho de que la ampolla aparezca sobre la piel sana. Aunque ello ocurre en la mayoría de las veces, este hecho no es constante y en ocasiones sólo se pone de manifiesto en las primeras horas de evolución de la ampolla. En otras la ampolla se rodea de un halo inflamatorio que puede ser más ostensible cuando el contenido de la ampolla se hace purulento (fig. 5).
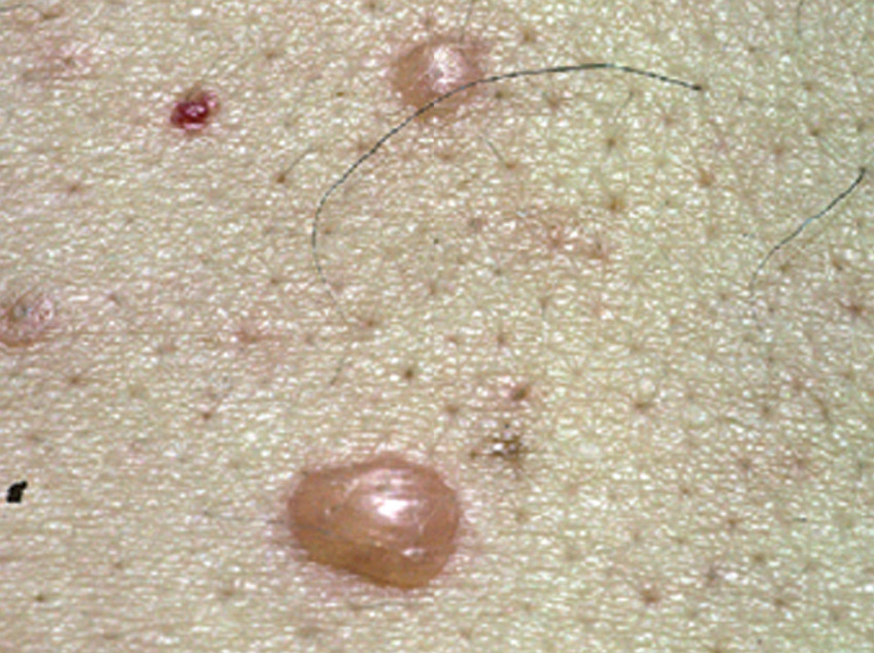
Fig. 2.--Ampolla transparente sobre piel sana.
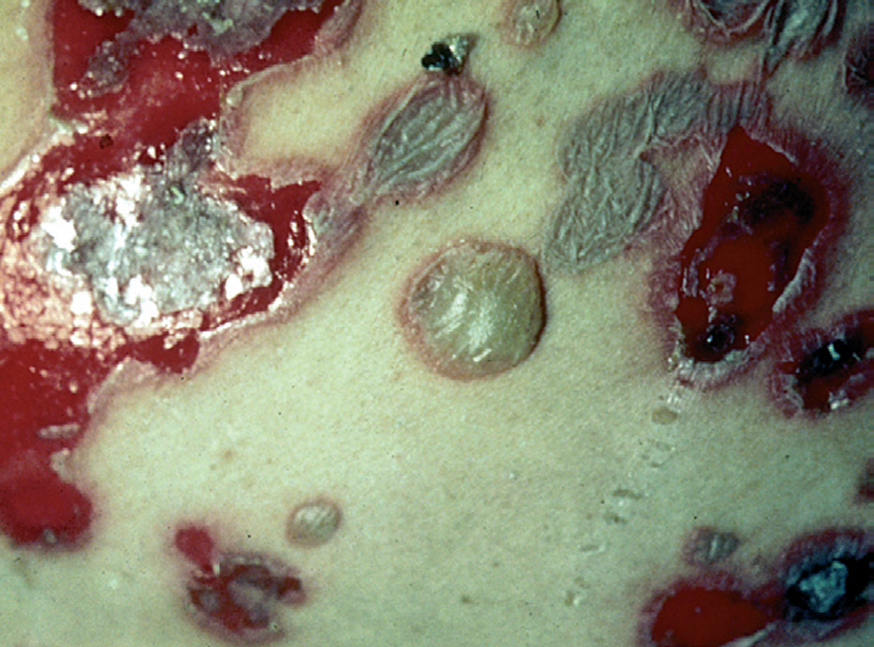
Fig. 3.--Ampollas flácidas, erosiones y costras.
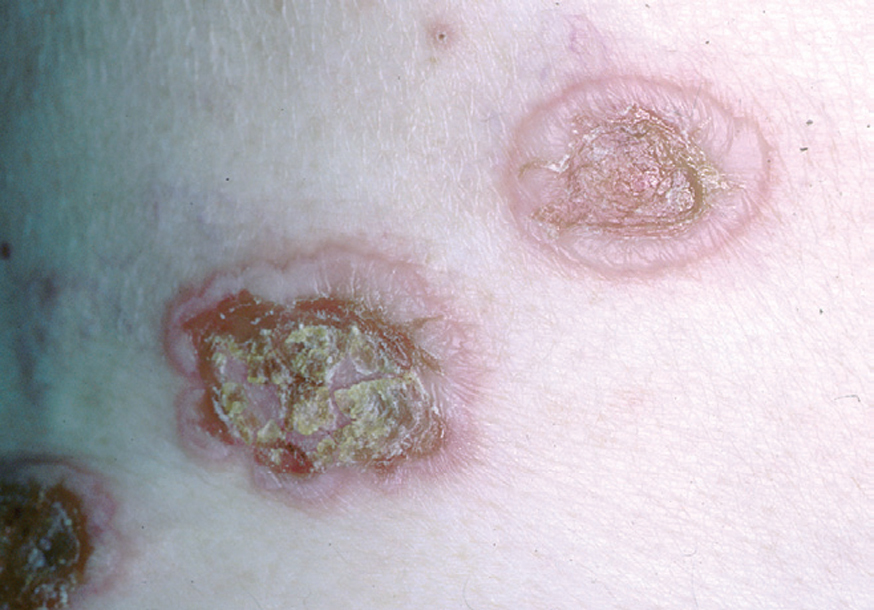
Fig. 4.--Extensión periférica de la ampolla.
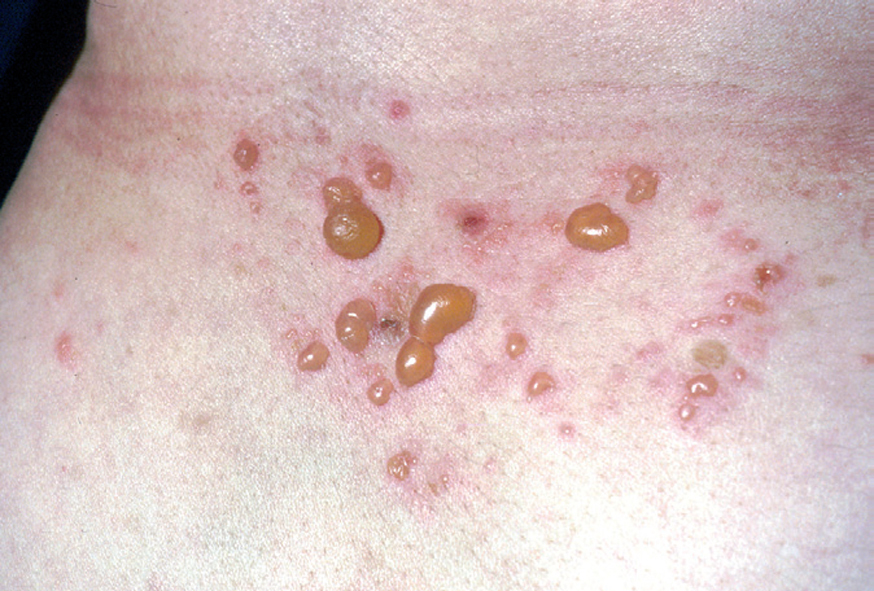
Fig. 5.--Ampollas agrupadas sobre un fondo eritematoso.
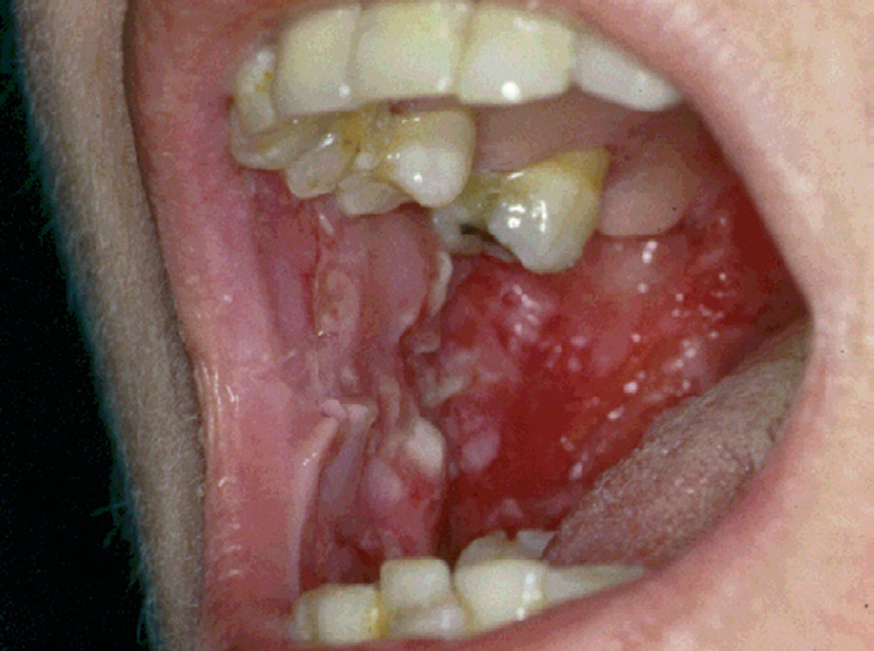
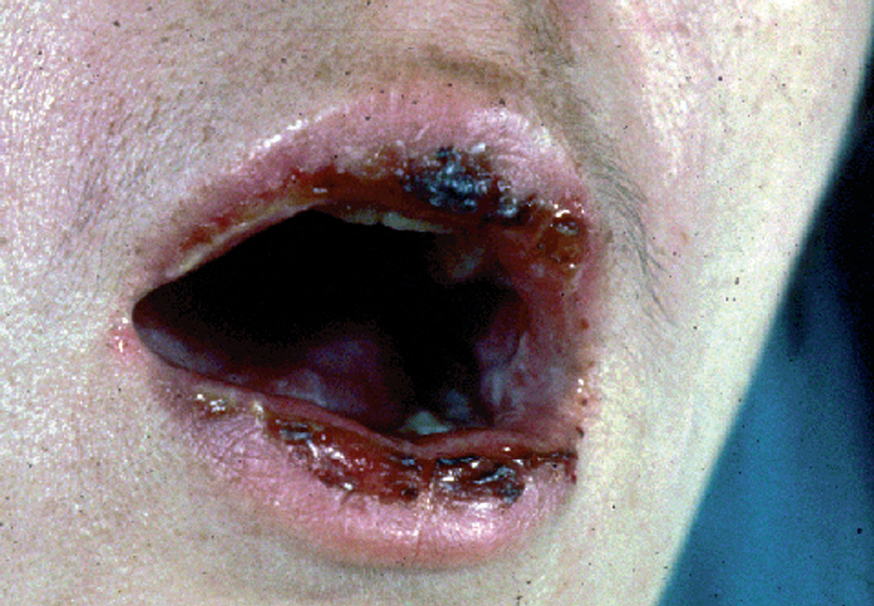
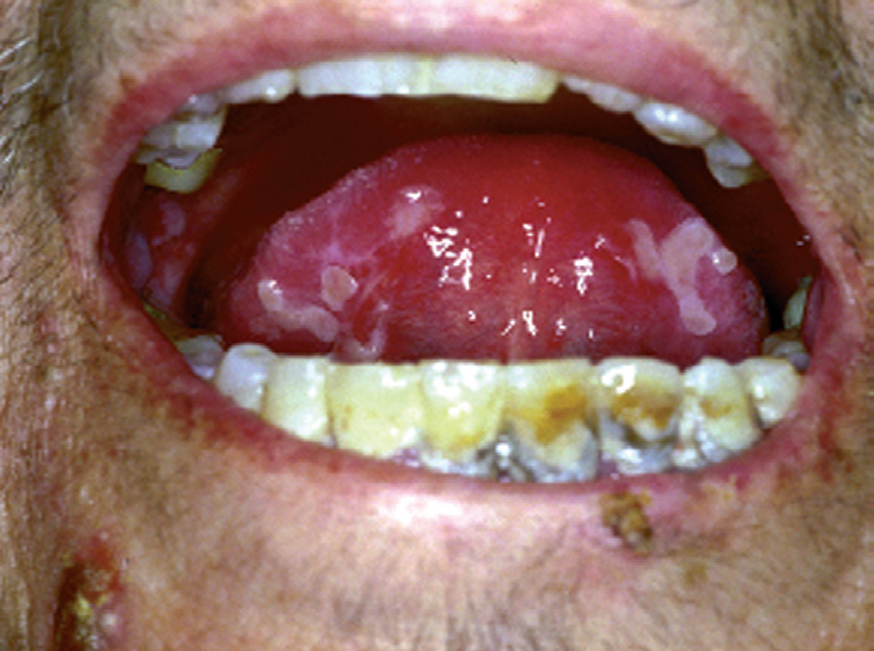
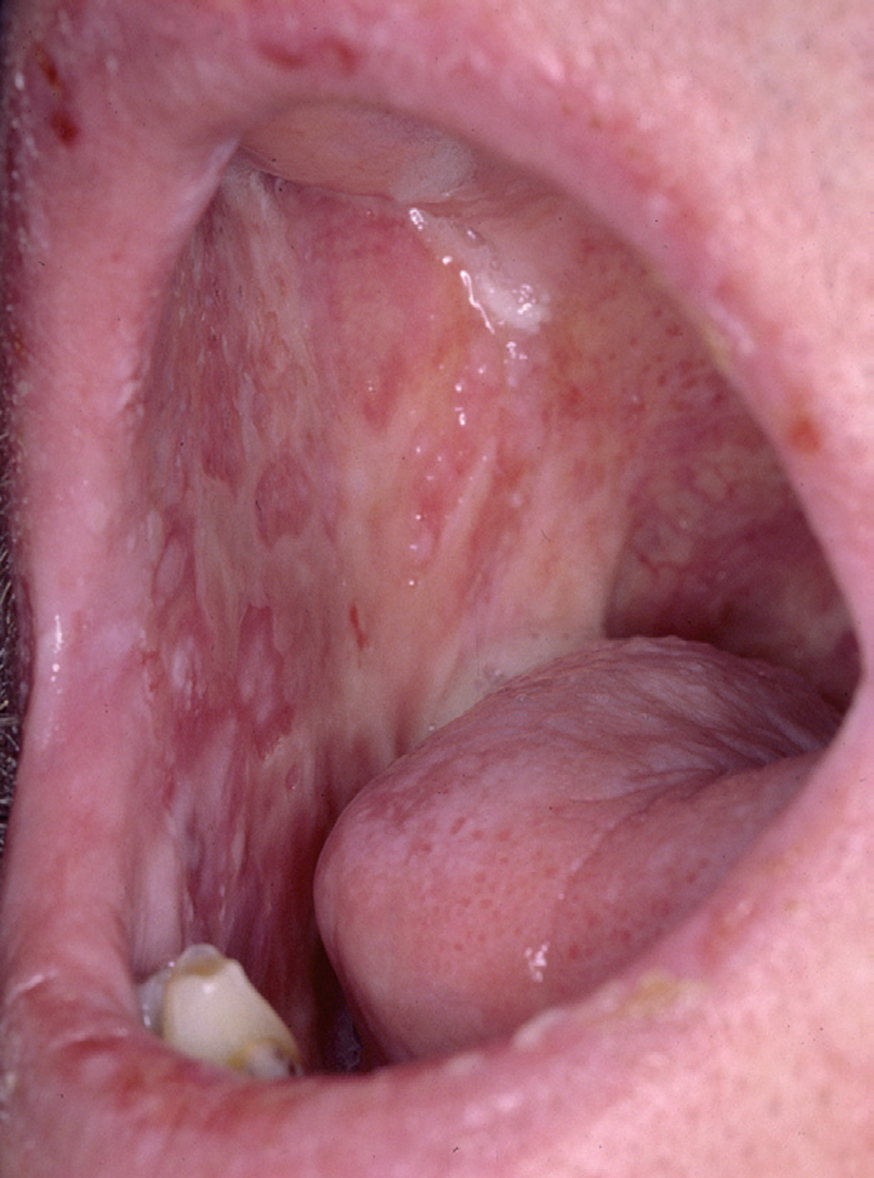
La forma de comienzo del cuadro ocurre en un porcentaje elevado de los casos, alrededor del 50-70 % de los mismos, con lesiones mucosas. La afectación mucosa es muy característica y frecuente, siendo un hecho casi constante que aparece en la evolución del PV. De ellas, la mucosa oral es la más frecuentemente interesada y, como ya hemos referido, en ocasiones marca el comienzo de la enfermedad. Estas lesiones suelen ser dolorosas y son resistentes al tratamiento. En la mucosa yugal las lesiones son muy evocadoras en forma de erosiones irregulares, grandes y extensas, que dejan al descubierto una mucosa hiperémica, o a veces cubiertas de lesiones blanquecinas (fig. 6). En los labios pueden existir erosiones y costras con un collarete descamativo o con restos epidérmicos (fig. 7). Cuando afecta a la mucosa gingival va a dar origen a erosiones alrededor de la implantación dentaria. No infrecuentemente se afecta el paladar tanto duro como blando en forma de erosiones aisladas o coalescentes, así como la lengua, en donde las lesiones erosivas pueden ser dolorosas (fig. 8). Otras mucosas, como la conjuntiva, la mucosa de la nariz u otras, pueden estar afectadas, ocurriendo en ocasiones la afectación de dos o más mucosas simultáneamente. En los casos en los que la enfermedad tiende a ser más grave se pueden encontrar lesiones en la faringe, laringe, esófago, mucosa anal y genital acompañados de dolor 119.
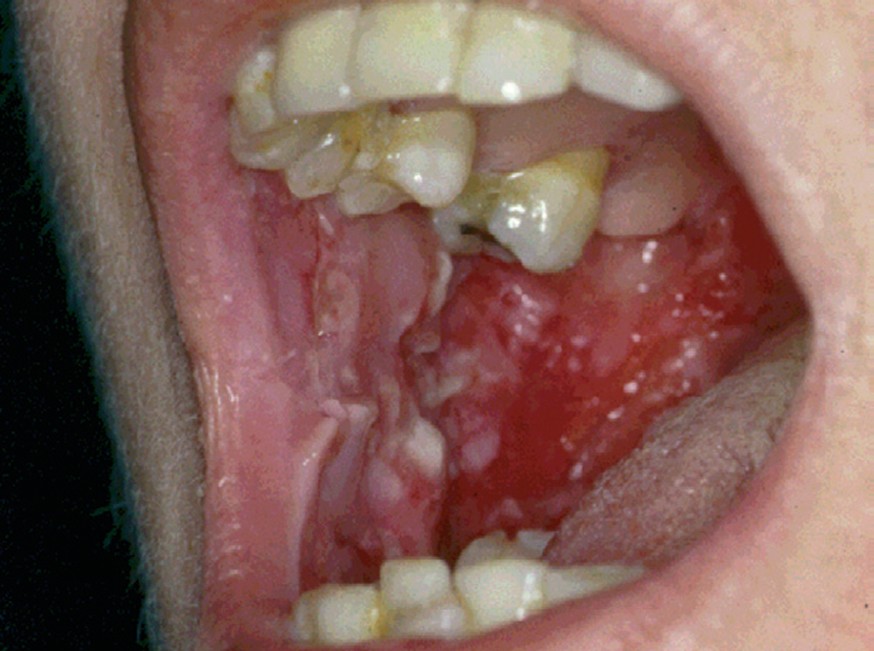
Fig. 6.--Erosiones en mucosa yugal en pénfigo vulgar.
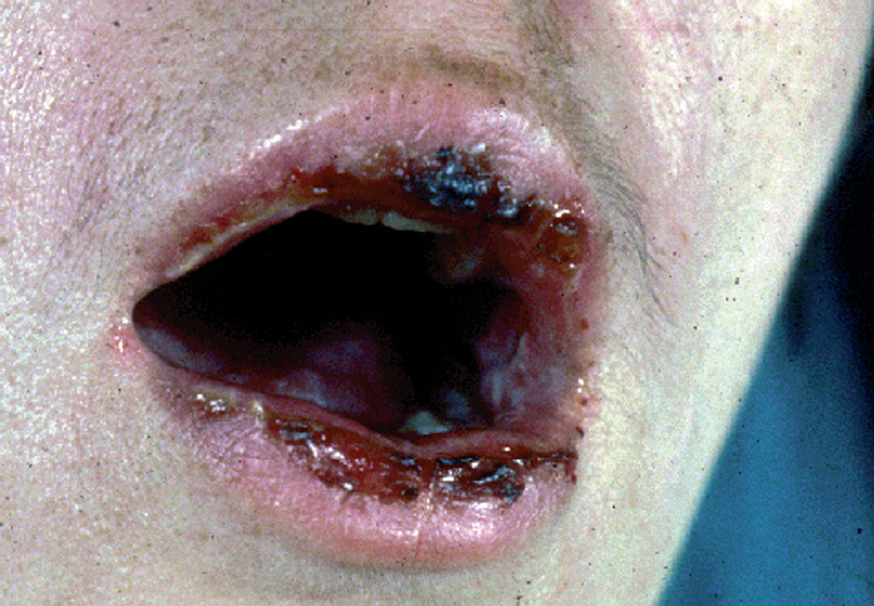
Fig. 7.--Queilitis erosiva y costrosa en pénfigo vulgar.
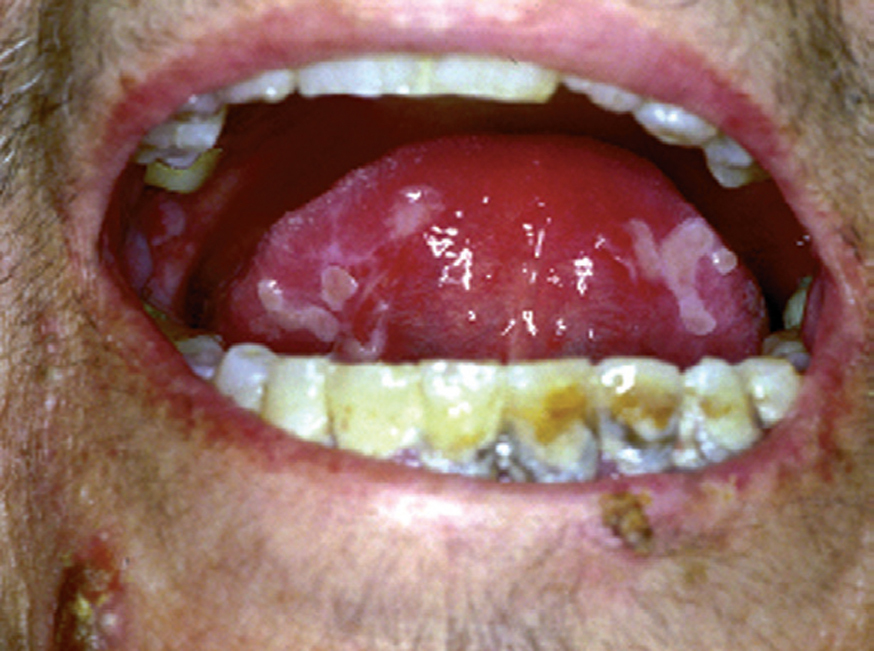
Fig. 8.--Lesiones erosivas y costrosas en lengua.
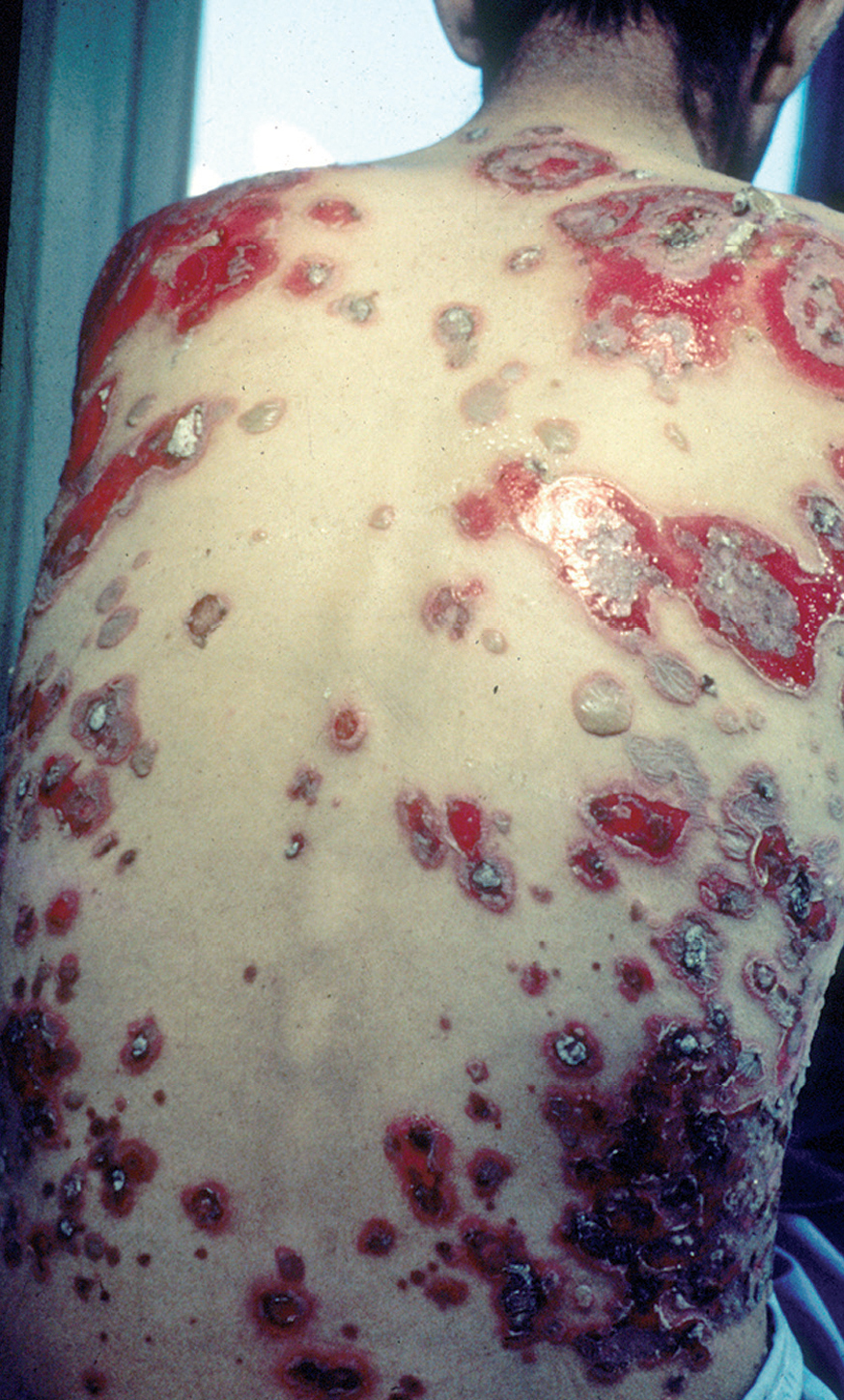
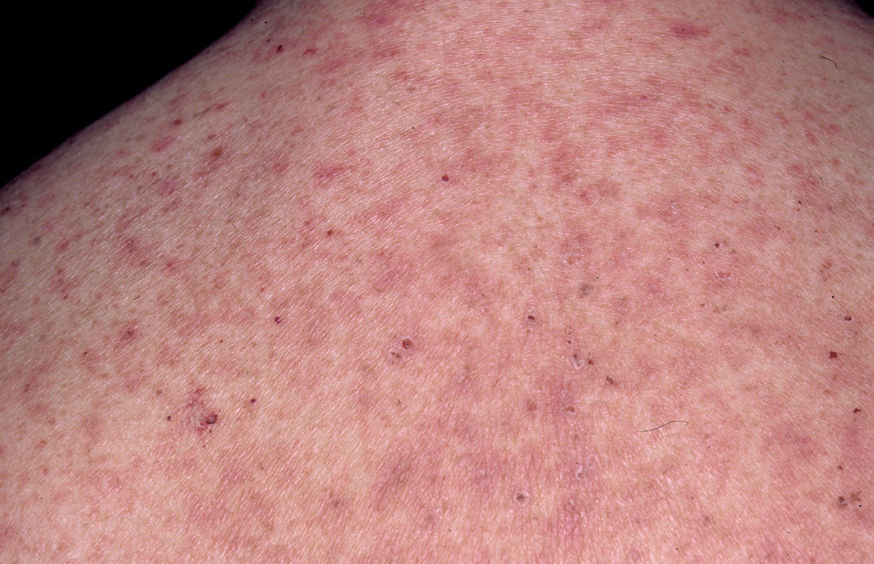
En el 10 o 15 % de los casos el PV puede iniciarse como manifestaciones cutáneas, bien como una ampolla única, que puede persistir un cierto tiempo, o como brote de lesiones múltiples más o menos diseminadas (fig. 9). Más frecuente es que las lesiones cutáneas aparezcan meses o años después de las lesiones mucosas y a veces de forma simultánea. Las ampollas pueden aparecer en cualquier parte de la superficie cutánea y muchas veces se disponen irregularmente sobre la piel y aparecen en brotes sucesivos, afectando con mayor frecuencia el cuero cabelludo, cara y axilas (fig. 10). Las ampollas, dada la fragilidad epidérmica suprayacente se desecan, pero más habitualmente se rompen, vertiendo el contenido líquido al exterior, siendo reemplazadas luego por erosiones, costras y eventualmente ulceraciones. Algunas al regresar pueden dejar una pigmentación residual. Esta mezcla de ampollas, costras, erosiones, láminas de epidermis despegada y lesiones pigmentadas configura el llamado falso polimorfismo de la lesión del pénfigo (fig. 11), debido a que de manera típica la única lesión elemental del pénfigo es la ampolla, siendo el resto de las lesiones el resultado de la evolución de ésta. Como hemos indicado, en el pénfigo existe una fragilidad epidérmica que desde el punto de vista clínico se pone de manifiesto con el signo de Nikolsky y el signo de Asboe-Hansen. El signo de Nikolsky consiste en la demostración del despegamiento epidérmico al hacer una presión tangencial con el dedo sobre la superficie de la piel. El signo de Asboe-Hansen consiste en el aumento periférico del tamaño de la ampolla al presionar verticalmente sobre la superficie de la misma. A pesar de la intensidad del cuadro clínico, existe característicamente en la piel una ausencia de manifestaciones subjetivas, singularmente del prurito. En ocasiones existe una sensación dolorosa de la piel no constante y, en algunos casos, las lesiones, si son erosivas o si asientan en el fondo de los pliegues, pueden ser dolorosas. Asimismo el estado general se encuentra conservado, por lo menos en las etapas precoces de la enfermedad.
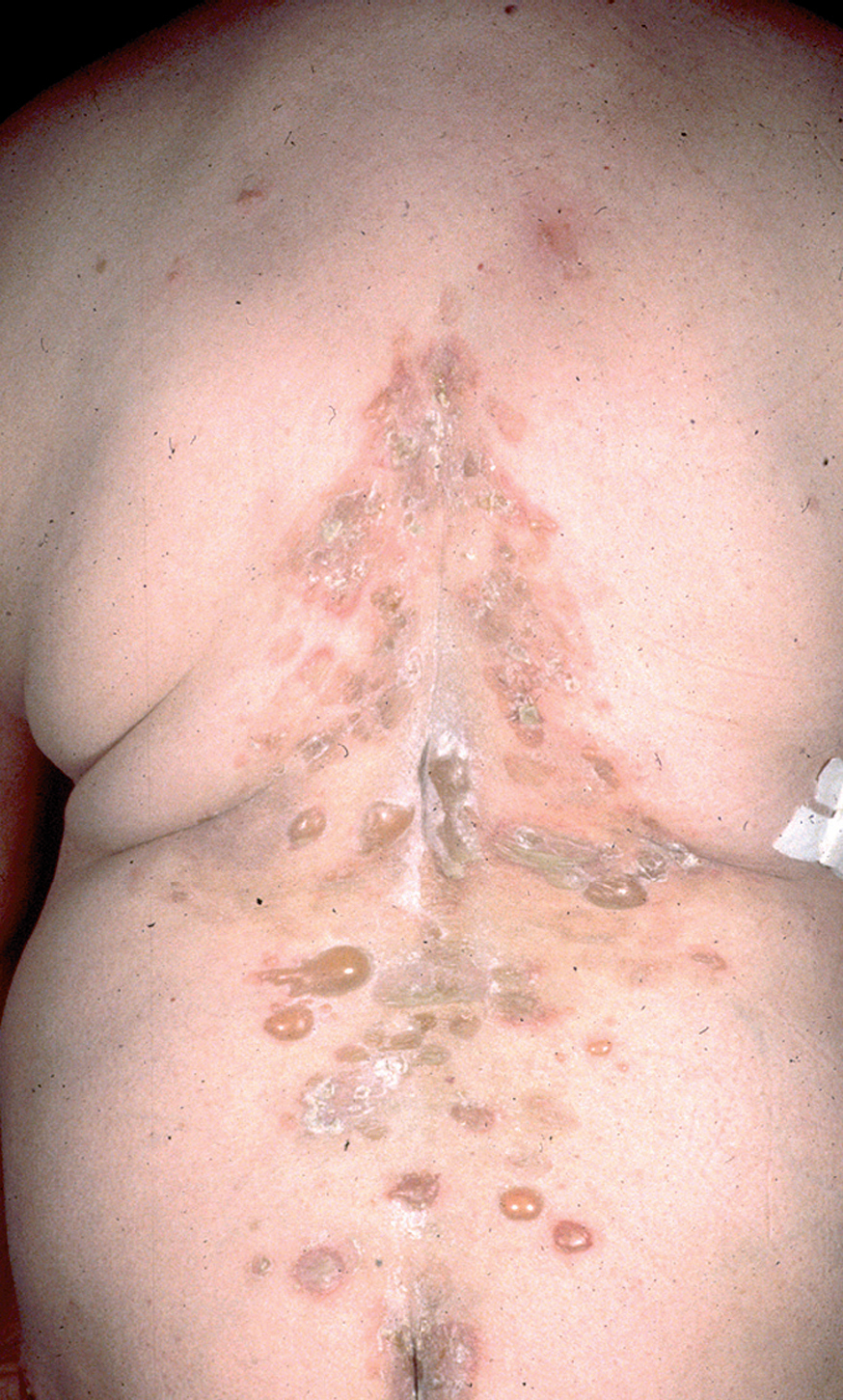
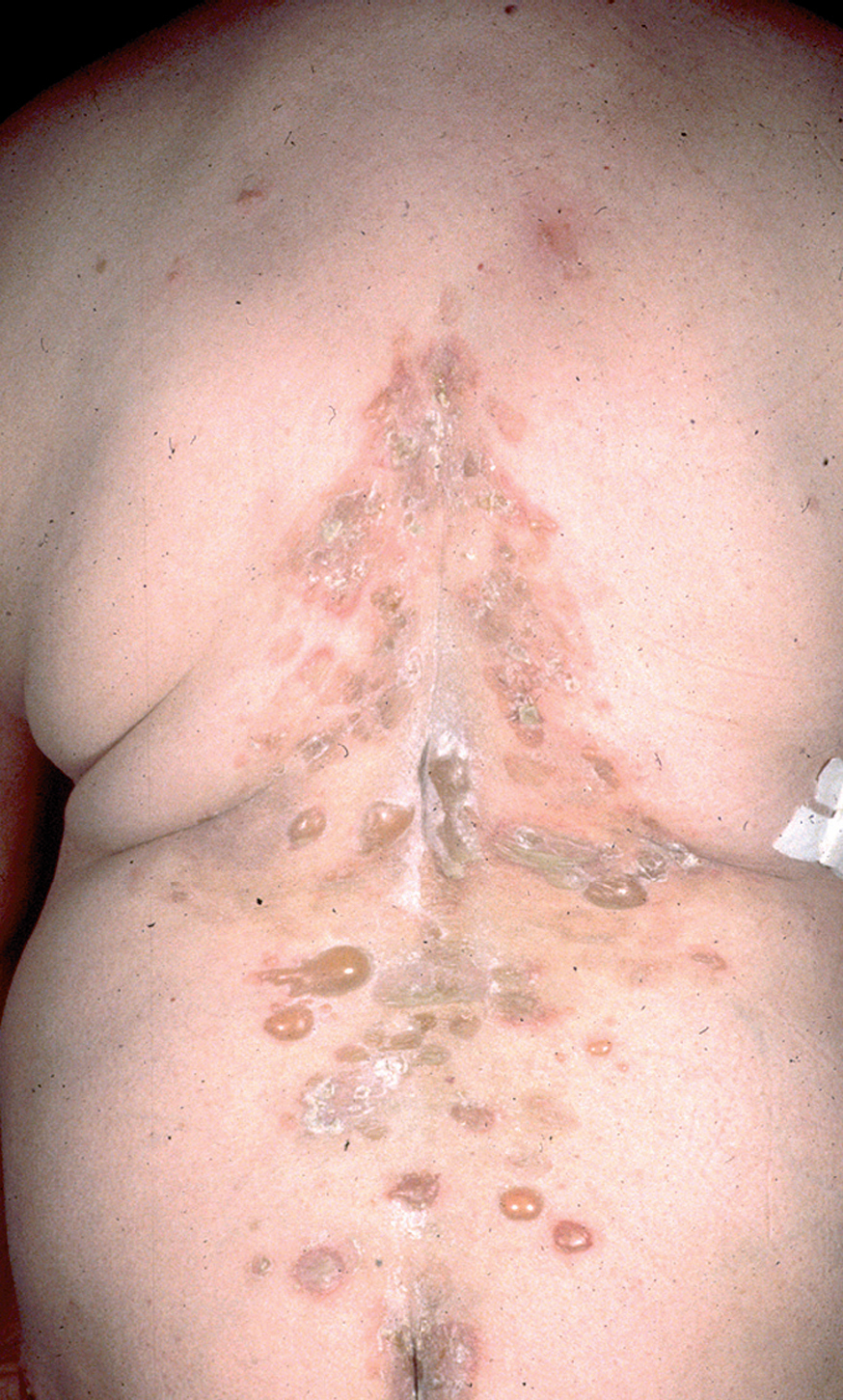
Fig. 9.--Ampollas en espalda de pénfigo vulgar.
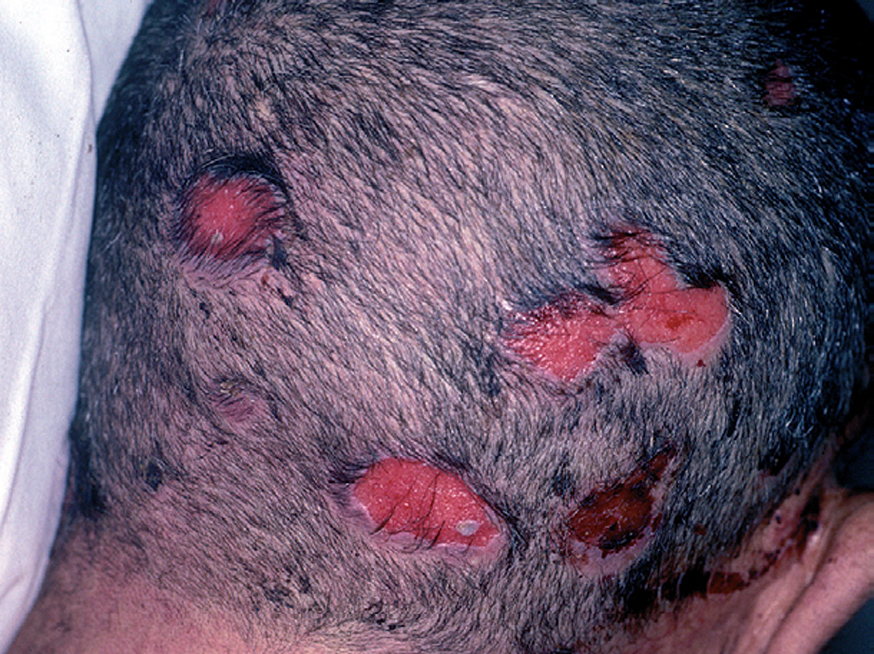
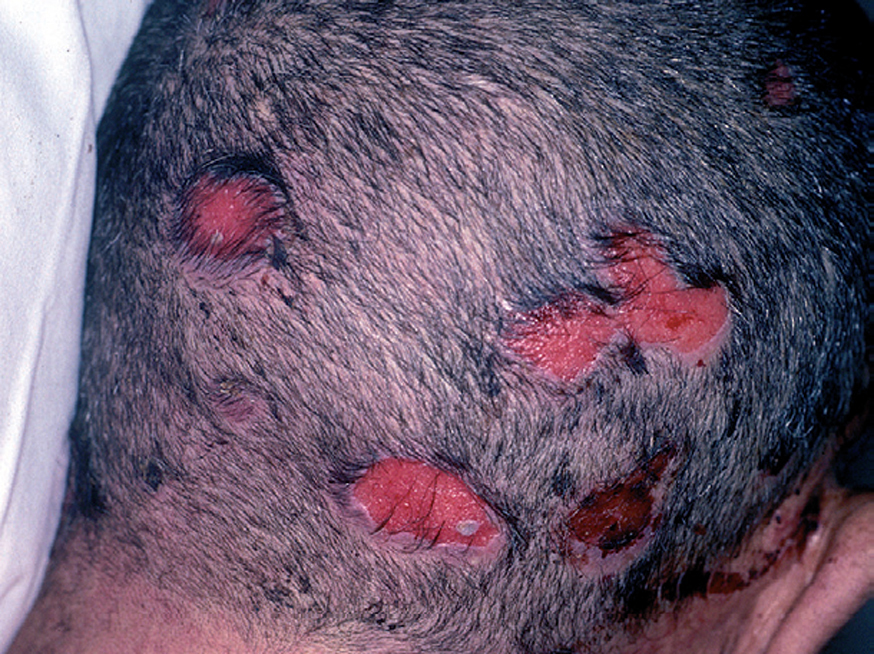
Fig. 10.--Lesiones erosivas en cuero cabelludo.
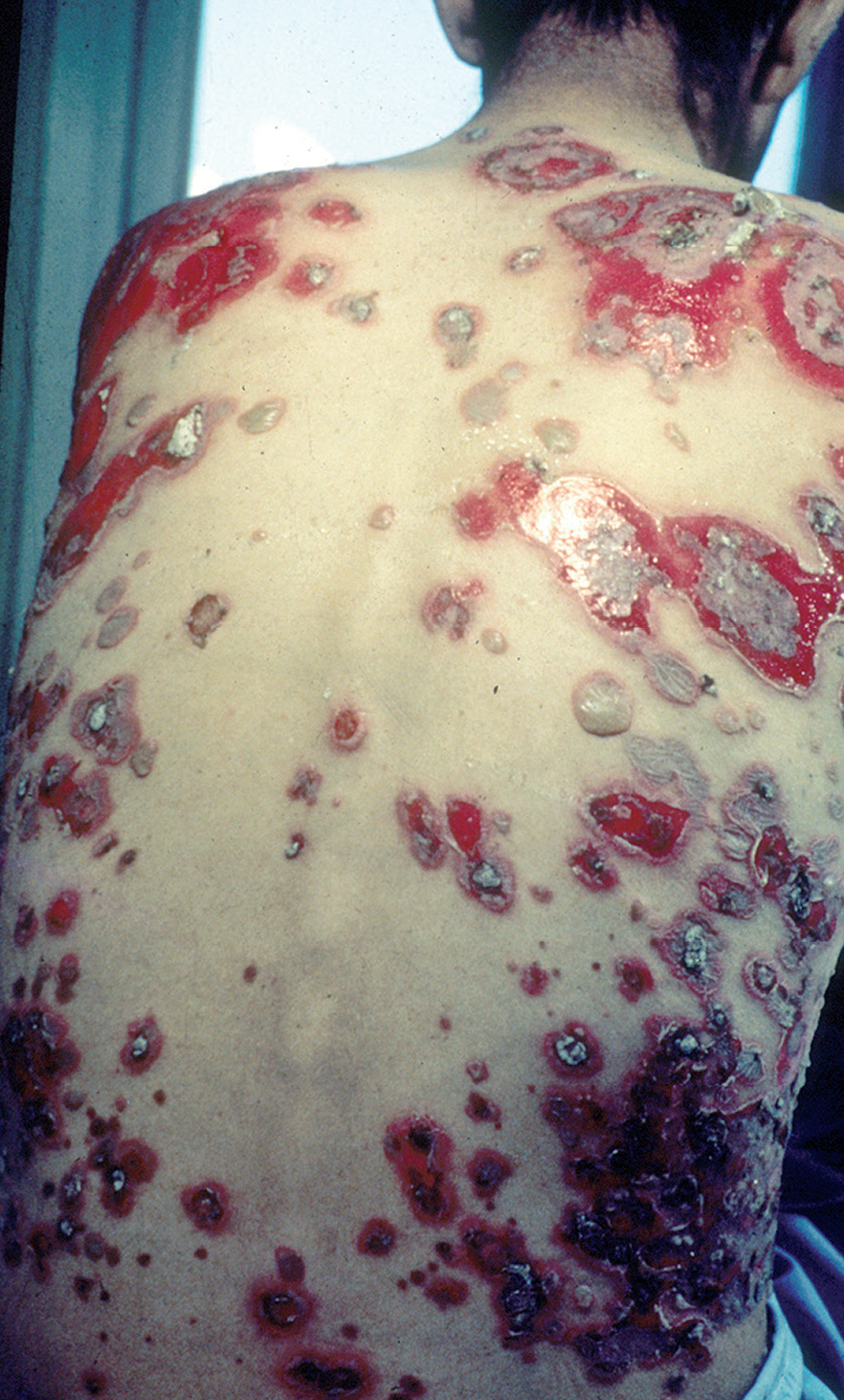
Fig. 11.--Falso polimorfismo del pénfigo vulgar.
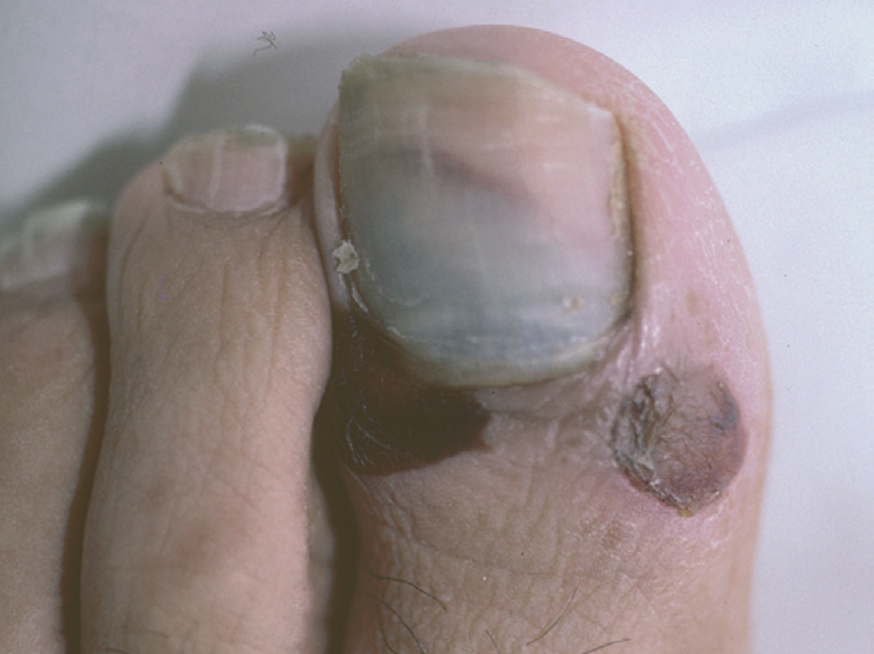
Las uñas pueden encontrarse afectadas junto con otras manifestaciones clínicas de pénfigo e incluso puede ser la primera manifestación de la enfermedad. Es un hallazgo clínico que aparece en el 22 % de los pacientes en una serie, con diversa morfología clínica como paroniquia, onicosquicia, onicomadesis, hemorragias subungueales (fig. 12), alteraciones de la coloración o aparición de líneas de Beau, que afectan sobre todo al 1.er y 2.º dedos de la mano, sin relación con la duración o gravedad de la enfermedad 120.
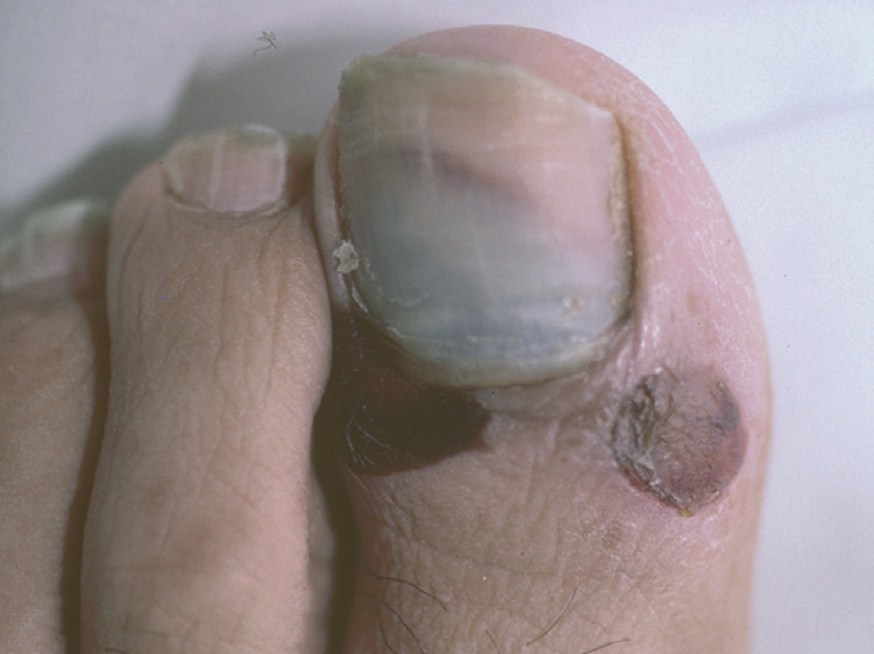
Fig. 12.--Hemorragia subungueal.
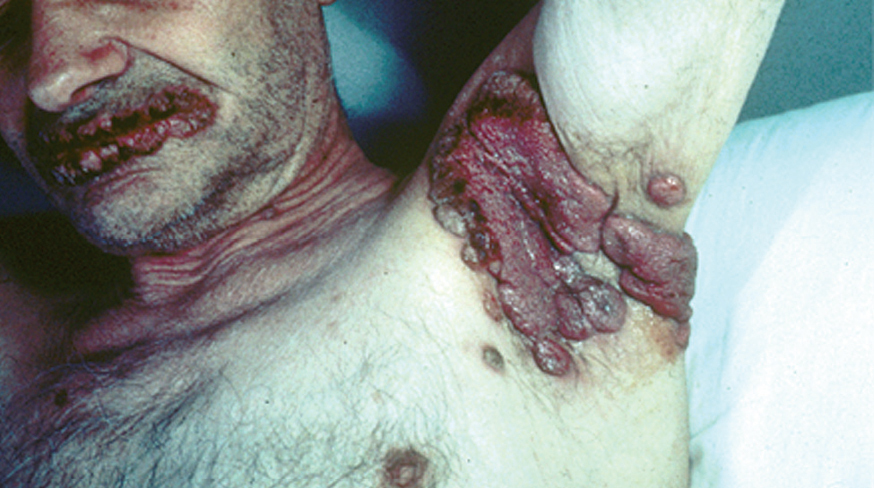
Los pénfigos vegetantes tipo Neumann y tipo Hallopeau son variantes clínicas del PV. Clínicamente presentan lesiones ampollosas que evolucionan a placas erosivas sobre las cuales aparecen lesiones vegetantes, con mal olor, de tamaño variable que, al evolucionar, por coalescencia, forman grandes placas vegetantes que en su periferia presentan ampollas o lesiones erosivas, localizados sobre todo en pliegues, cara y cuero cabelludo (figs. 13 y 14) Hoy se tiende a considerar que ambos subtipos de pénfigo, vegetante y vulgar, representan un espectro clínico que va desde la forma más grave, que es el PV, una forma intermedia que es el pénfigo vegetante de Neumann y la forma más benigna que es el pénfigo vegetante de Hallopeau con remisiones espontáneas en ocasiones 121.
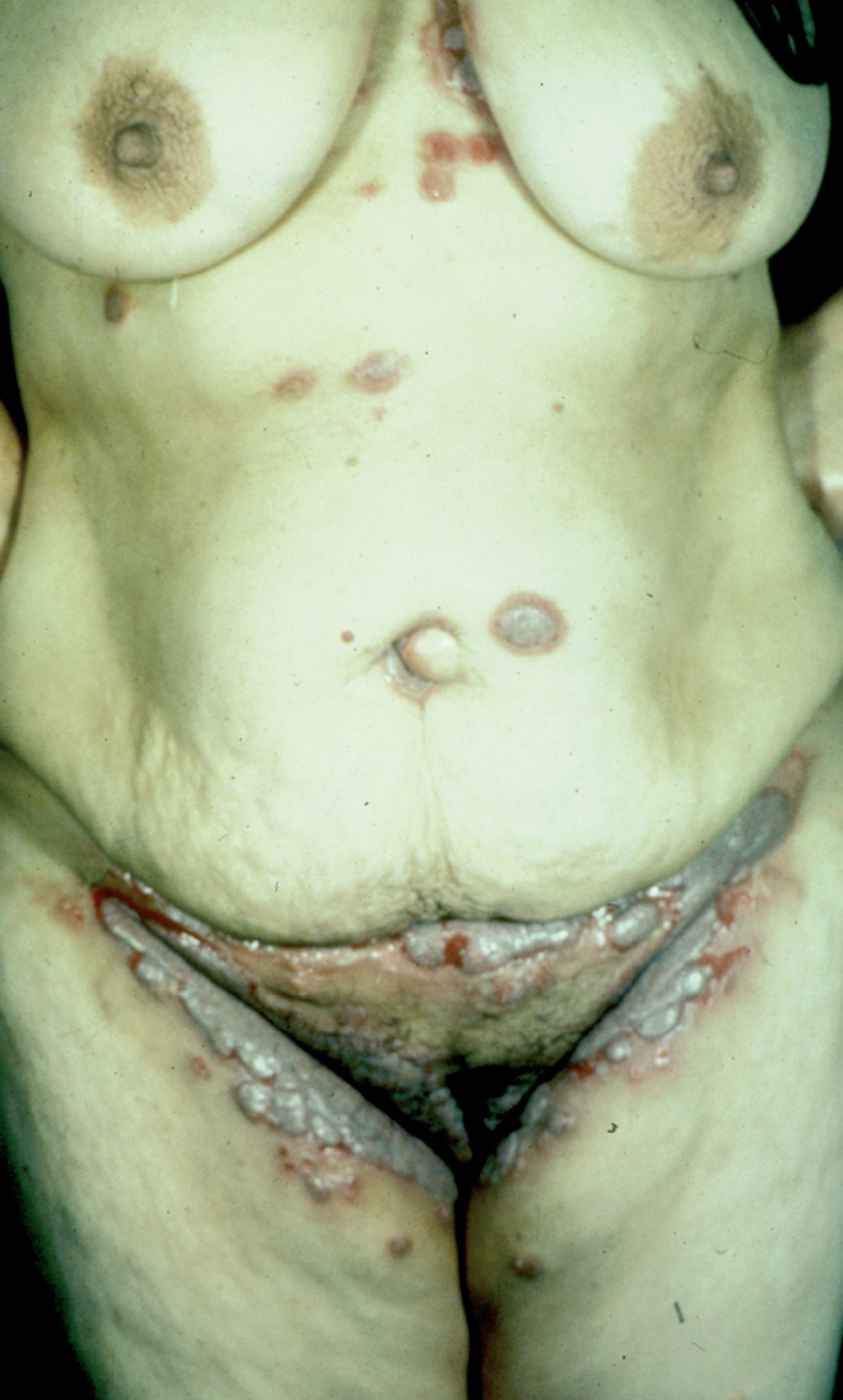
Fig. 13.--Pénfigo vegetante.
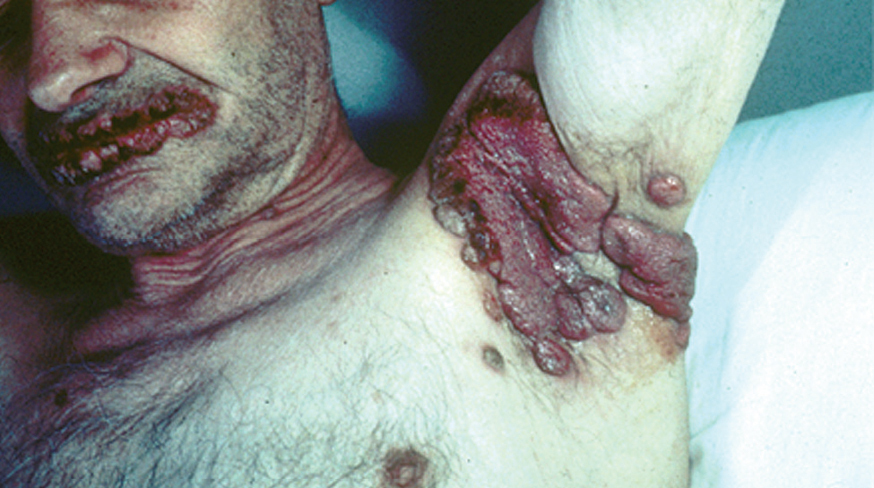
Fig. 14.--Pénfigo vegetante.
De forma excepcional, pacientes con hallazgos clínicos típicos, histopatológicos e inmunológicos de PV evolucionan a PF con síntomas, histopatología e inmunología característica 122-125. debido al fenómeno de difusión del epítopo. Se ha descrito la asociación de miastenia grave y/o timoma con pénfigo en la literatura médica, aunque es poco frecuente 126.
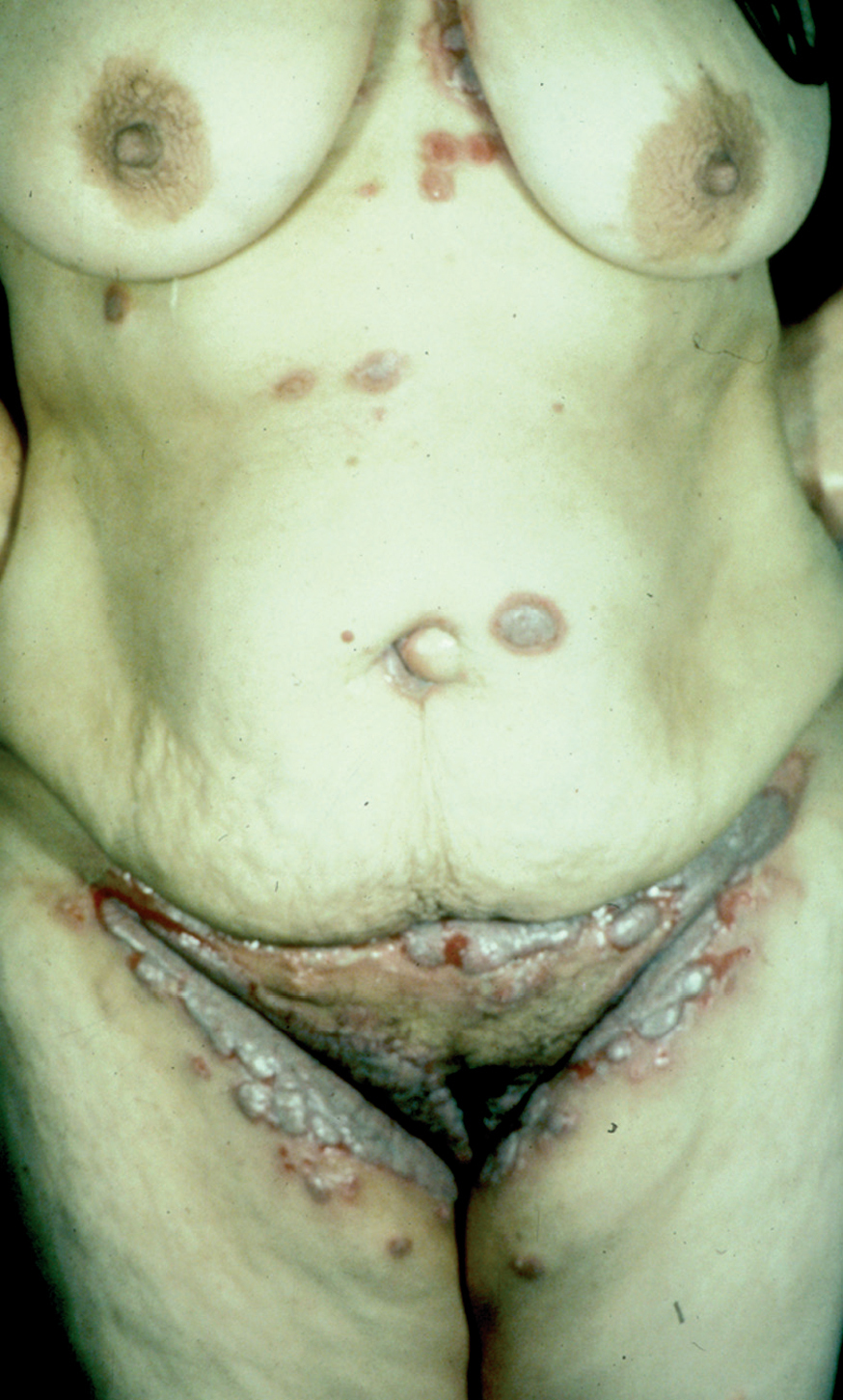
Pénfigo foliáceo
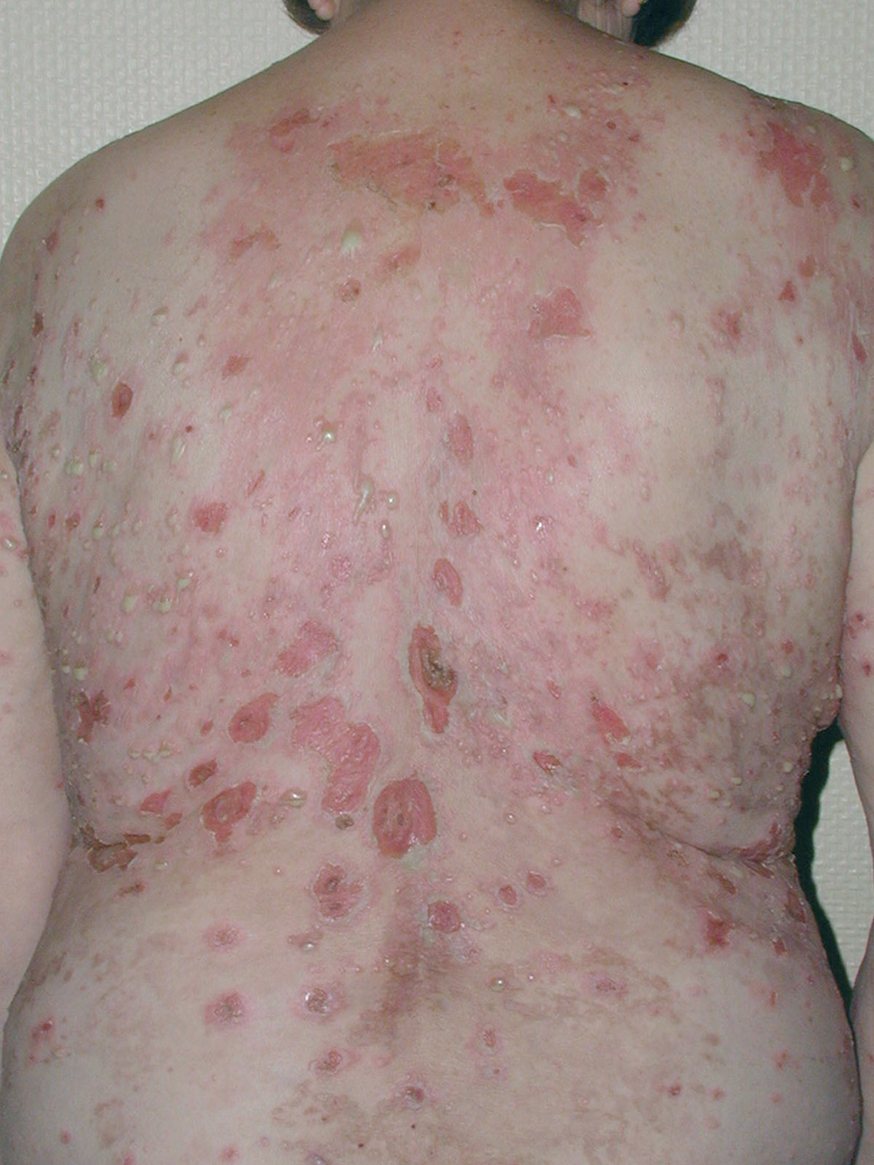
El cuadro de PF no endémico se caracteriza inicialmente por la presencia de ampollas localizadas superficialmente sobre una base discretamente eritematosa, y por lo tanto desde el punto de vista clínico son frágiles y fugaces, por lo que rara vez se observan, siendo reemplazadas por erosiones exudativas y costrosas, rodeadas de eritema. La fragilidad epidérmica puede ponerse de manifiesto con el signo de Nikolsky.
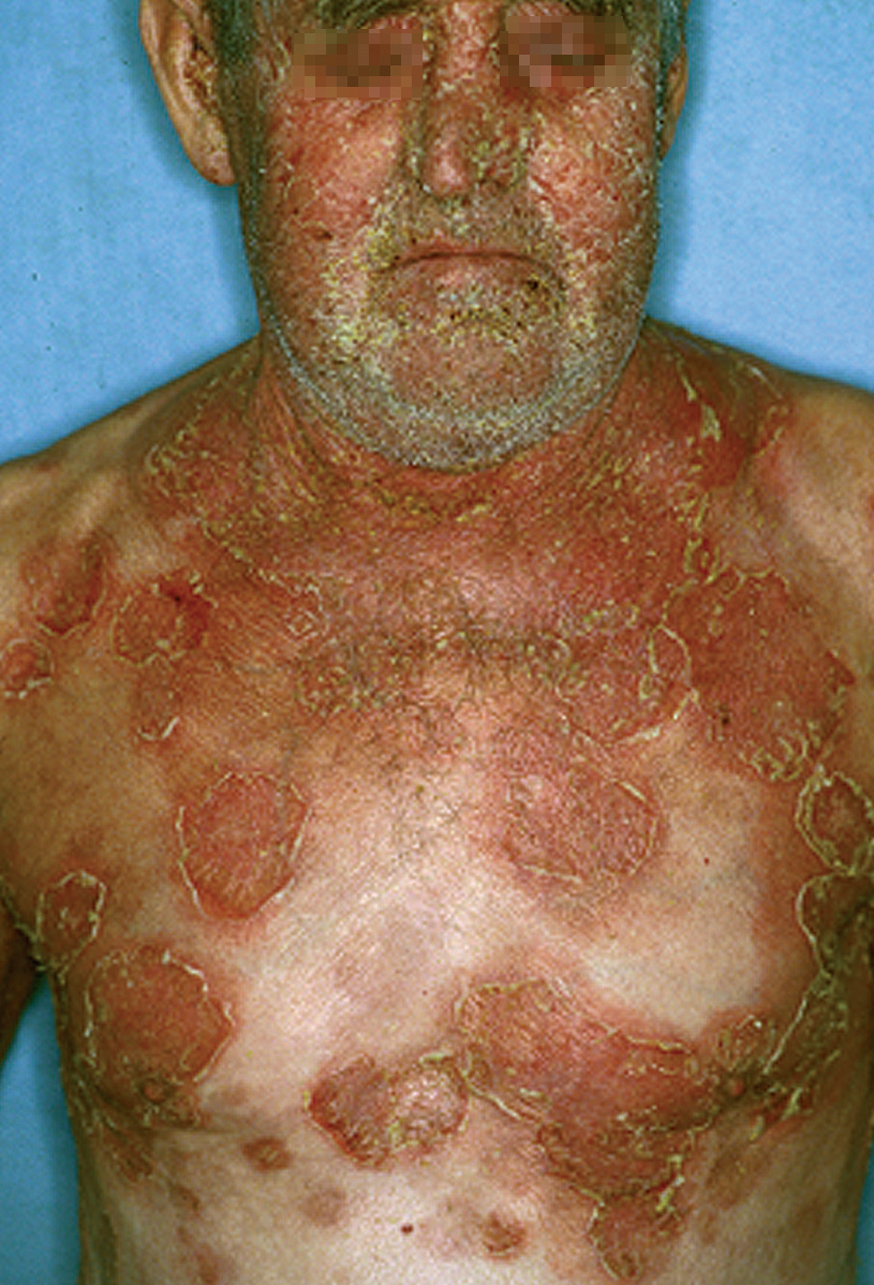
Las lesiones inicialmente se localizan en cuero cabelludo, cara, cuello, tórax y espalda, aunque puede aparecer en cualquier región de la superficie corporal (fig. 15). Las lesiones pueden estar limitadas a estas zonas, pero en la mayoría de los casos se diseminan en semanas o meses, por la confluencia de lesiones previas y aparición de nuevas lesiones. Las erosiones pueden diseminarse dando lugar a una eritrodermia (fig. 16). Es característico que la mucosa oral esté respetada. El cuadro clínico habitualmente es crónico.
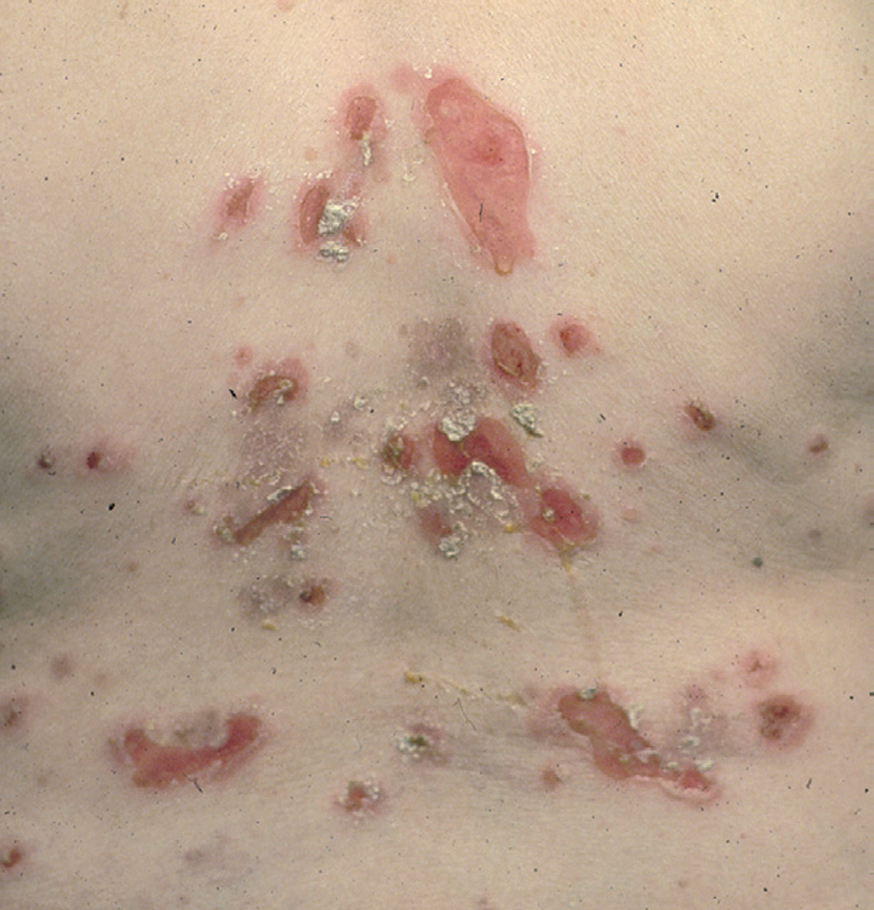
Fig. 15.--Placas erosivodescamativas en pénfigo foliáceo.
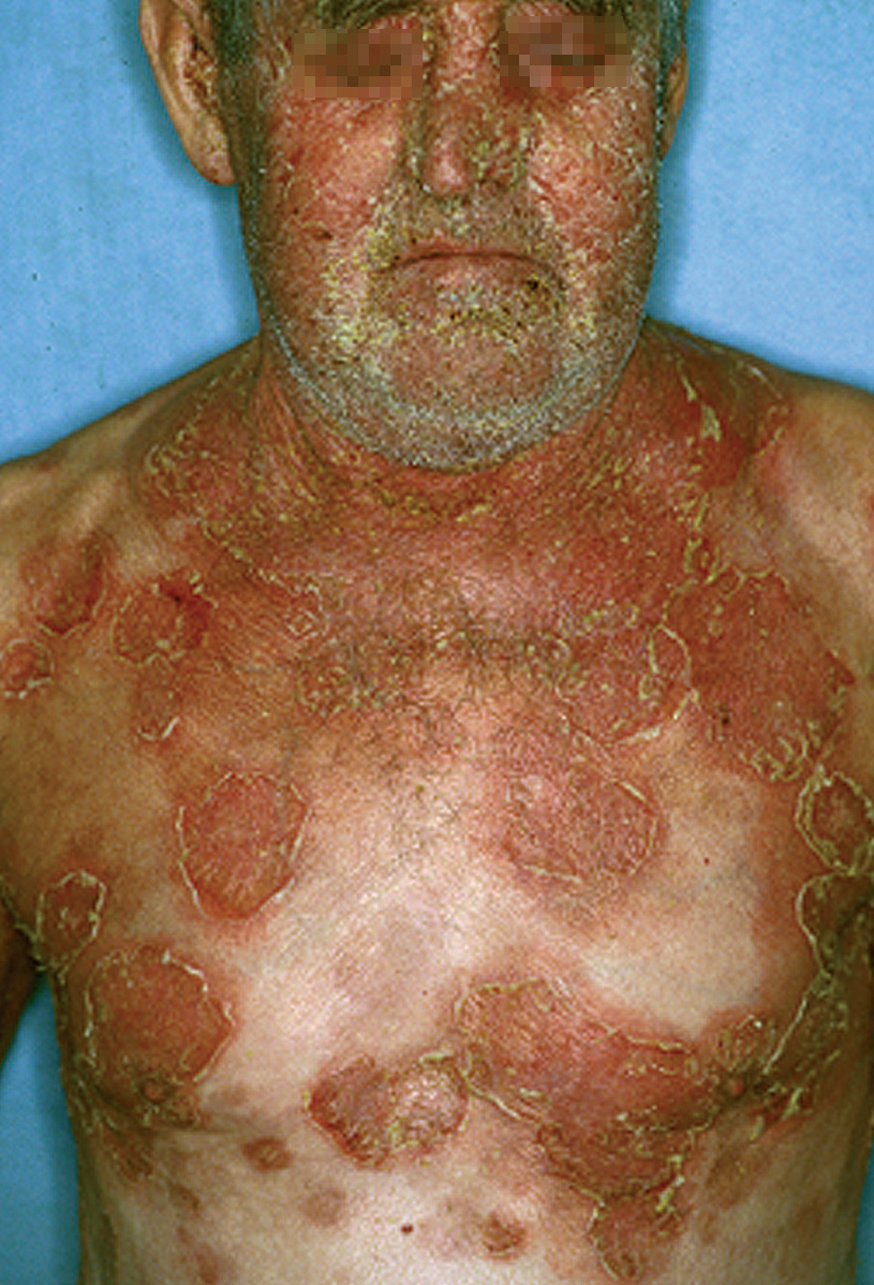
Fig. 16.--Pénfigo foliáceo.
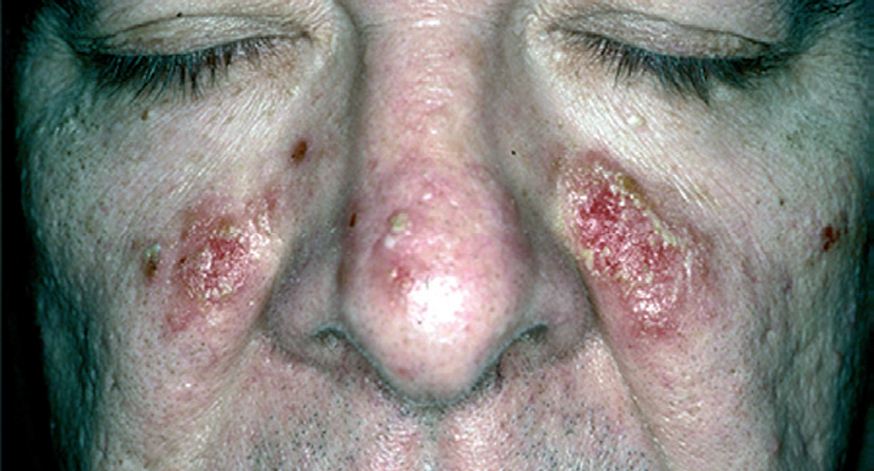
El pénfigo eritematoso es una variante clínica localizada de PF con placas eritematosas descamativas y costrosas, habitualmente en la región centrofacial y otras veces en la región submamaria o preesternal que recuerdan a las lesiones de lupus eritematoso (fig. 17). En un porcentaje que oscila entre el 30 y el 80 % se observan en el suero anticuerpos antinucleares. El PF o eritematoso es la forma de presentación clínica más frecuente en el pénfigo inducido por medicamentos del grupo tiol, como la D-penicilamina (fig. 18).
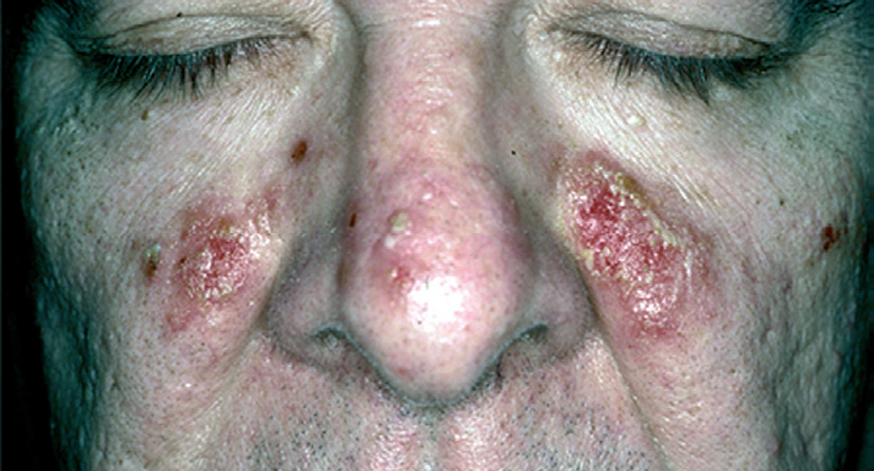
Fig. 17.--Pénfigo eritematoso.
Fig. 18.--Pénfigo foliáceo inducido por D-penicilamina.
El fogo selvagem es el PF endémico, clínica, histológica e inmunológicamente indistinguible del PF no endémico. Es frecuente en Brasil, en áreas rurales, y afecta a jóvenes, sospechándose como factor desencadenante un factor ambiental hasta ahora desconocido. El cuadro clínico más frecuente consiste en placas eritematosas descamativas diseminadas sobre todo en áreas seborreicas y en zonas expuestas a la luz UV, simulando el aspecto de una quemadura. Pueden observarse otros patrones clínicos como la forma ampollosa-exfoliativa, la eritrodermia, placas queratósicas o verrugosas, forma semejante a la dermatitis herpetiforme y formas hiperpigmentadas, asociadas estas últimas a una remisión de la enfermedad 127.
En ocasiones pacientes con hallazgos clínicos típicos, histopatológicos e inmunológicos de PF evolucionan a PV con sintomatología, histopatología e inmunología característica 124.
HISTOPATOLOGIA
Es muy importante que la ampolla de la piel seleccionada para la biopsia sea de reciente aparición y, si es posible, se realizará una biopsia escisional. En mucosas puede ser difícil encontrar una ampolla intacta; en estos casos es mejor realizar la biopsia de una zona aparentemente sana de mucosa contigua a un área erosionada.
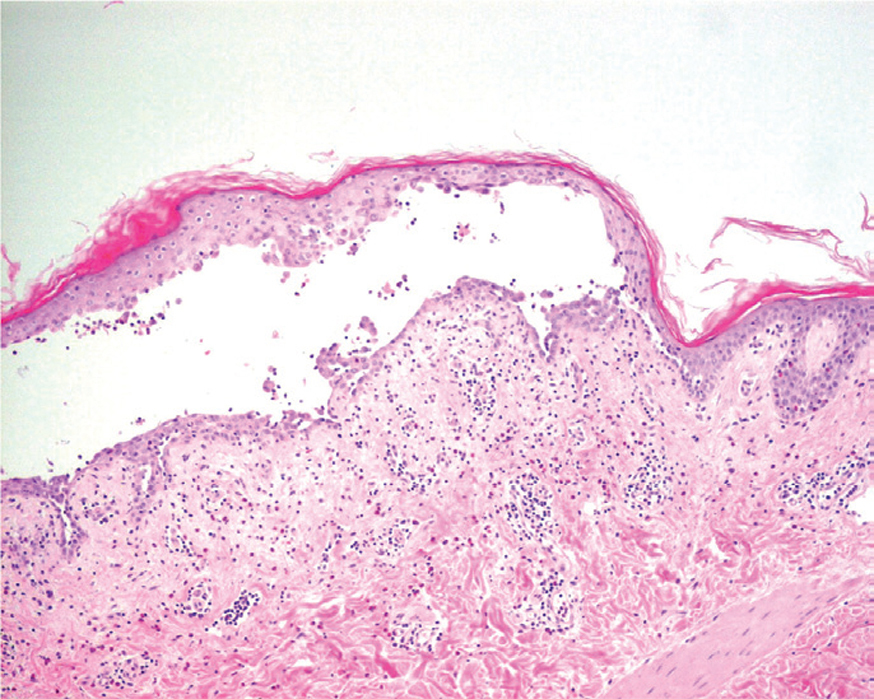
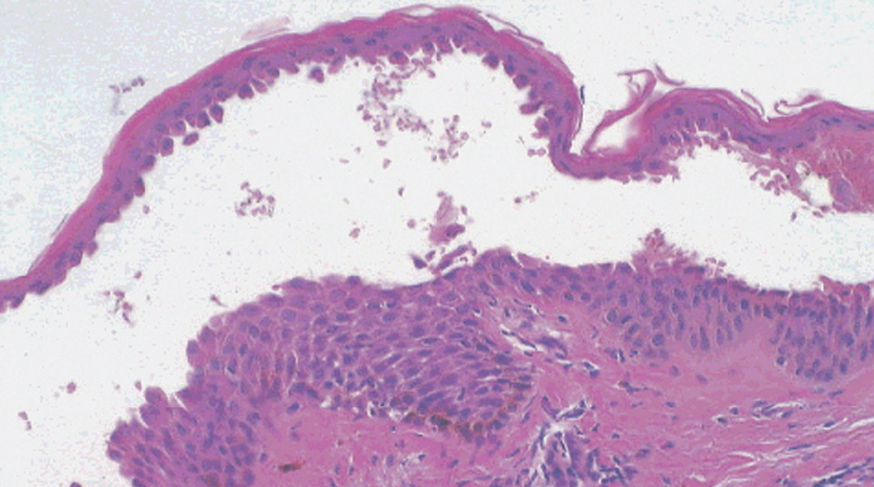
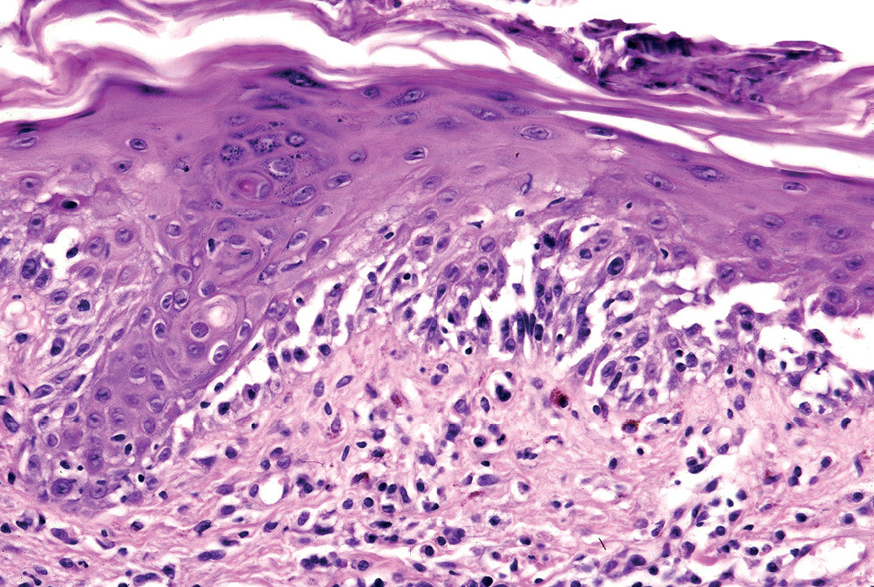
La primera alteración que se observa en la histopatología del PV es un edema intercelular que se localiza en las capas más inferiores de la epidermis, con desaparición de los puentes intercelulares, conduciendo así al fenómeno de acantolisis. Cuando la acantolisis progresa da lugar a la ampolla intraepidérmica suprabasal cuyo suelo está formado por una hilera de células basales, que se ha dicho que se disponen como una «hilera de lápidas sepulcrales», y cuyo techo se encuentra constituido por el resto de la capa espinosa, las capas granulosas y la córnea (fig. 19). En el interior de la ampolla se suelen encontrar queratinocitos acantolíticos, bien de manera aislada o en grupos, con una apariencia característica, cuya morfología es redondeada en lugar de poligonal, con un núcleo pequeño e hipercromático, con frecuencia rodeado de un halo y citoplasma homogéneo.
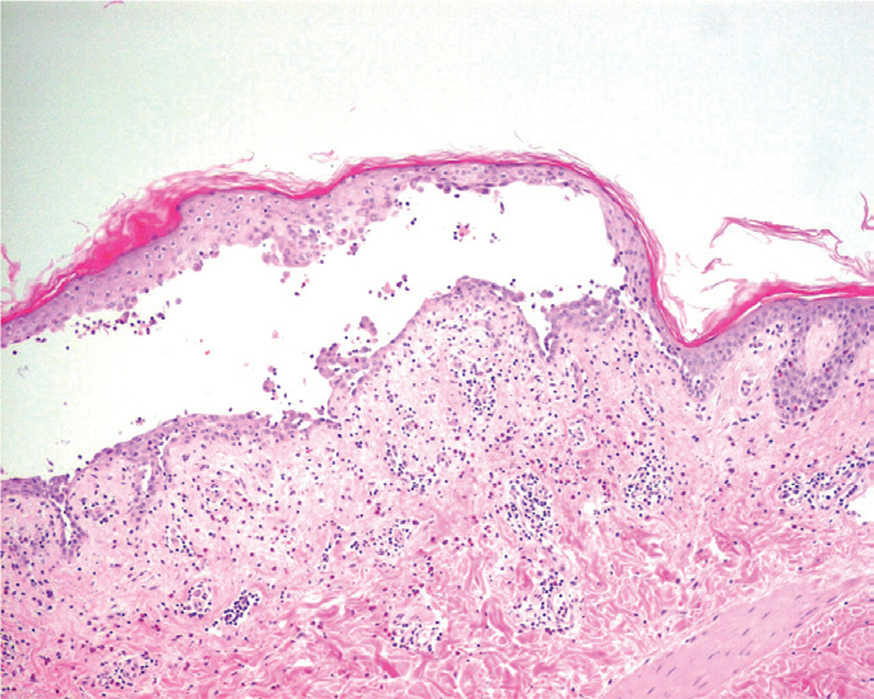
Fig. 19.--Ampolla suprabasal de pénfigo vulgar.
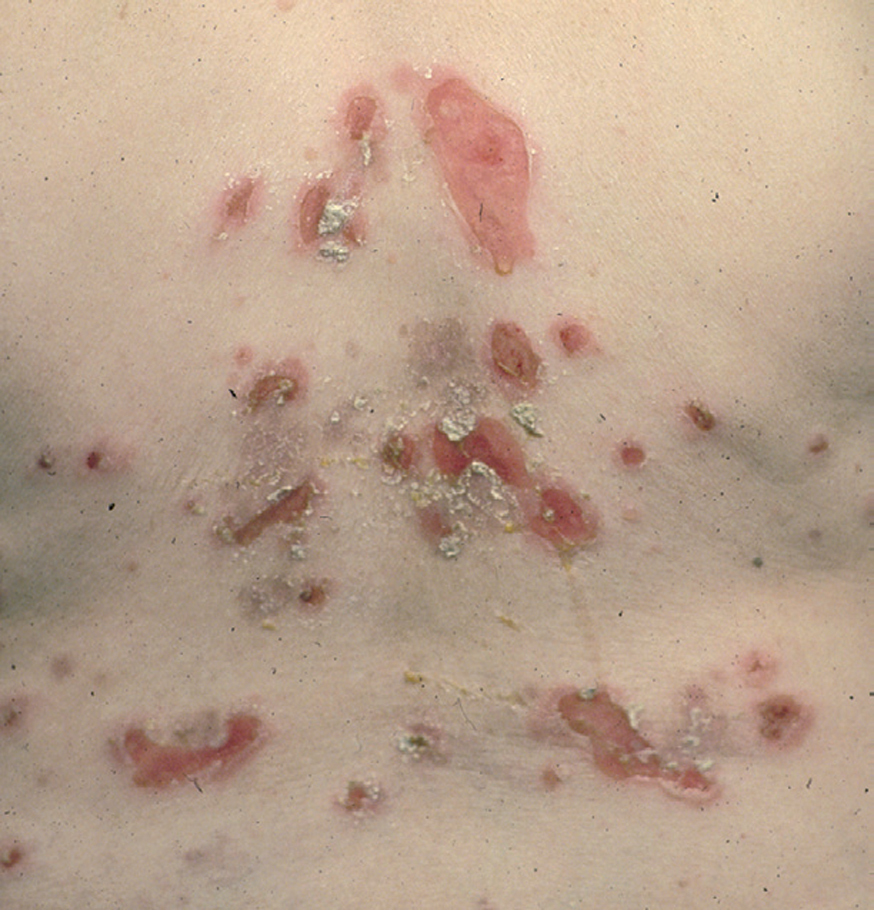
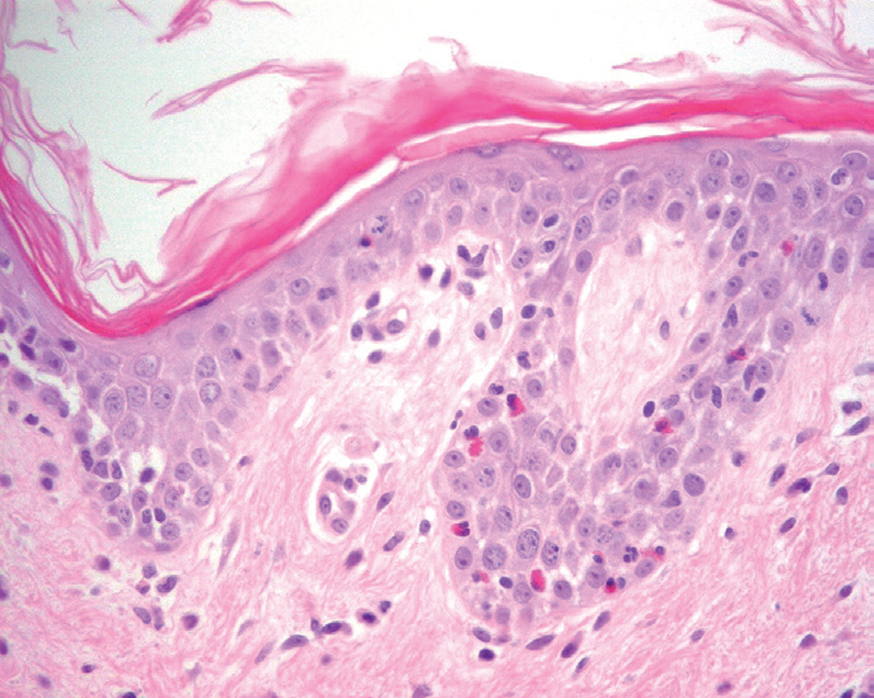
Más infrecuentes el hallazgo histopatológico inicial es una espongiosis eosinofílica (fig. 20) y neutrofílica, a veces en la misma biopsia. La hendidura puede afectar no sólo a la epidermis, sino también a los anejos, de manera que se puede observar en la totalidad del folículo terminal. También pueden encontrarse implicadas la glándula sudorípara y la glándula sebácea.
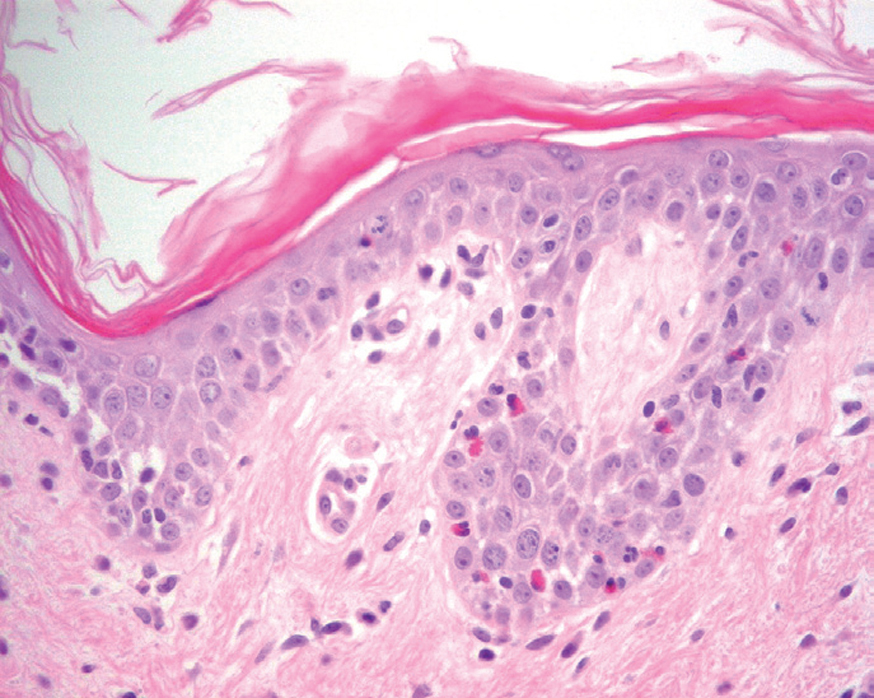
Fig. 20.--Espongiosis eosinofílica.
El proceso inflamatorio en la dermis es escaso en las fases precoces y queda limitado a un infiltrado linfocítico perivascular, acompañado de edema dérmico. En ocasiones el número de eosinófilos en dermis es elevado, acompañando a la espongiosis eosinofílica. Más tarde el infiltrado inflamatorio dérmico es mixto con neutrófilos, linfocitos, macrófagos y eosinófilos.
El pénfigo vegetante presenta acantolisis suprabasal, hiperqueratosis con costras escamosas, papilomatosis y abscesos intraepidérmicos en el que predominan los eosinófilos.
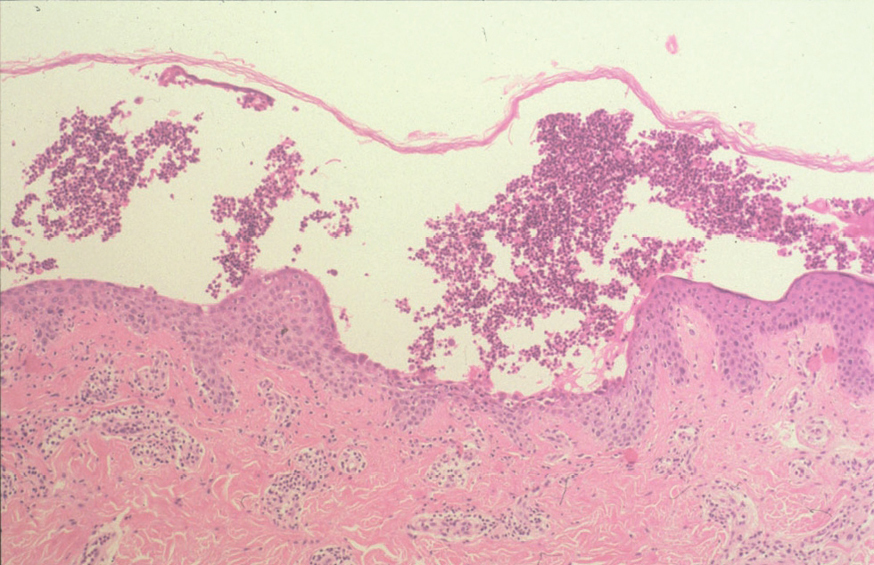
El PF y el eritematoso pueden mostrar un patrón de espongiosis eosinofílica, una ampolla subcórnea con escasos queratinocitos acantolíticos que evolucionan a queratinocitos granulares disqueratósicos característicos de esta enfermedad (fig. 21). El infiltrado inflamatorio observado es variable, dependiendo de la antigüedad de la lesión, el tipo de lesión biopsiada y de la existencia de impetiginización 128.
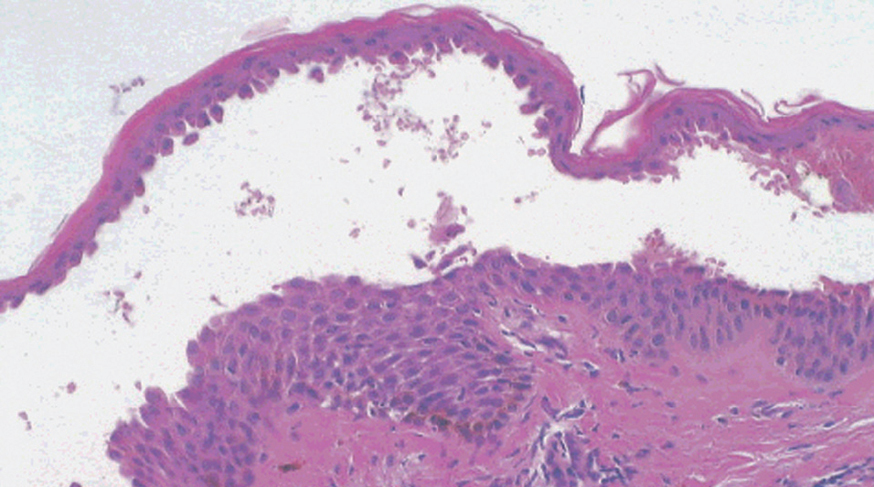
Fig. 21.--Ampolla intraepidérmica superficial en pénfigo foliáceo.
INMUNOFLUORESCENCIA DIRECTA
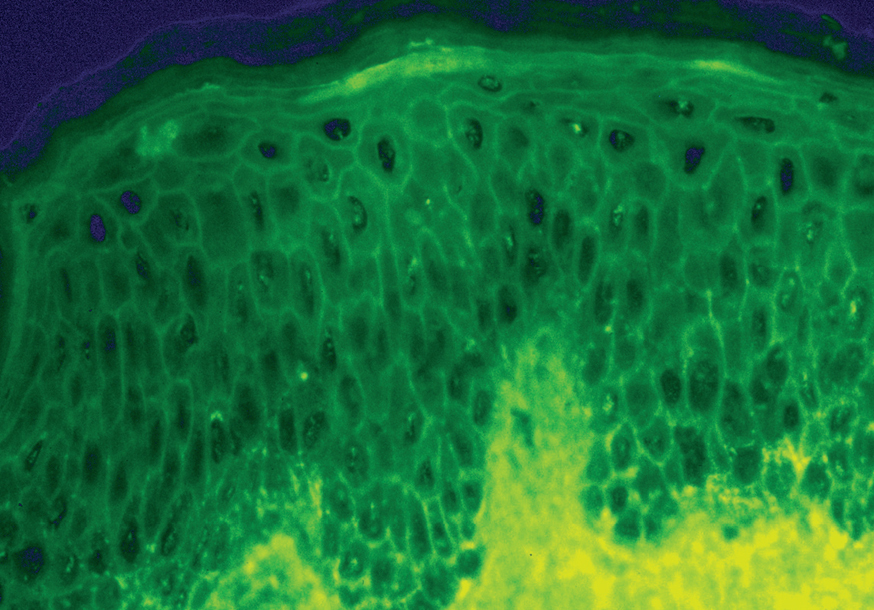
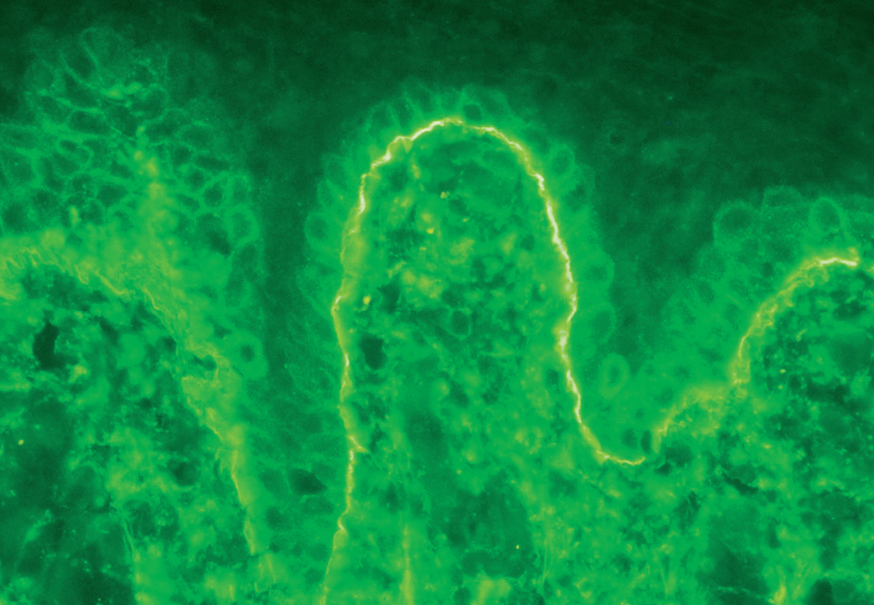
La demostración del depósito intercelular de IgG alrededor de los queratinocitos por inmunofluorescencia directa en piel perilesional es una prueba sensible y específica que confirma el diagnóstico de pénfigo (fig. 22). En el PV el depósito de anticuerpos es suprabasal. En el PF los depósitos se observan en el estrato granuloso y en la porción superior de la capa espinosa, y en otras ocasiones se demuestran en toda la epidermis. Con frecuencia la fracción C3 del complemento se encuentra en estas localizaciones. La inmunofluorescencia directa se puede utilizar como un marcador muy fiable de la remisión del pénfigo 129,130.
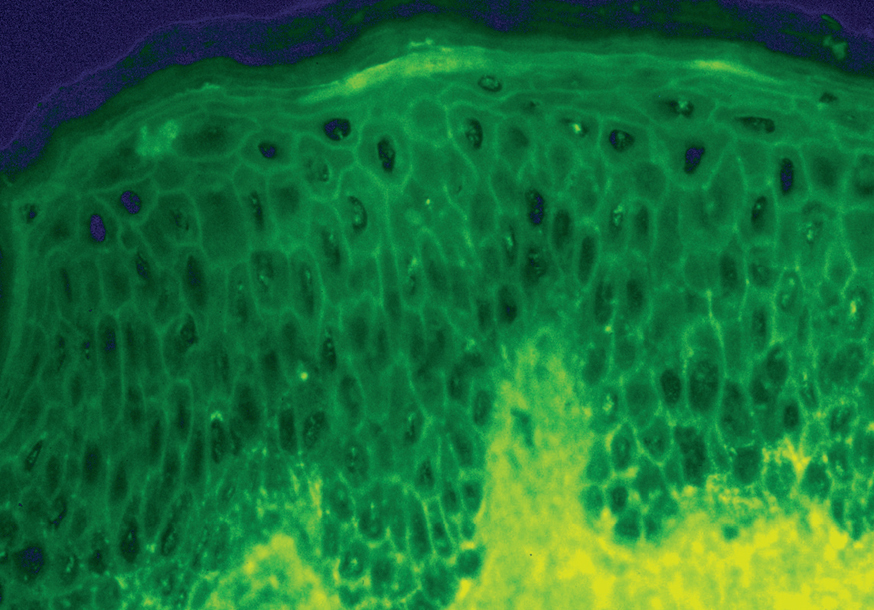
Fig. 22.--Depósito de IgG intercelular en pénfigo vulgar.
El pénfigo eritematoso combina el patrón anterior con depósito lineal-granular de inmunoglobulinas y/o fracciones del complemento en la unión dermoepidérmica, que sugiere la participación de mecanismos inmunopatológicos adicionales 131.
INMUNOFLUORESCENCIA INDIRECTA
La inmunofluorescencia indirecta confirma la existencia de anticuerpos circulantes, habitualmente IgG en el suero de la mayoría de los enfermos en piel humana o mucosa animal. En el PV el sustrato con mayor porcentaje de sensibilidad es el esófago de mono, mientras que en el PF es el esófago del cerdo de guinea 132.
El título de anticuerpos IgG circulantes del suero de la mayoría de los enfermos está en relación con la extensión y con la actividad de la enfermedad, y es un marcador evolutivo que, junto con la remisión clínica, se utiliza para valorar la eficacia del tratamiento 133.
TÉCNICA ELISA
La síntesis de desmogleínas 1 y 3 recombinantes en su estado conformacional ha permitido el desarrollo de una técnica de ELISA para la detección de anticuerpos antidesmogleína con una sensibilidad y especifidad al menos similar a la inmunofluorescencia indirecta. Es una técnica que detecta los antígenos frente a los que van dirigidos los autoanticuerpos, siendo útil posiblemente para monitorizar la actividad de la enfermedad 134-137. Tiene las ventajas sobre la inmunofluorescencia indirecta de ser una técnica sencilla, objetiva y que a veces ofrece resultados positivos cuando la IFI es negativa 138. Sin embargo, la técnica de ELISA no es útil para cuantificar los anticuerpos en sueros con niveles elevados de éstos 139, por lo que se ha propuesto realizar una dilución previa del suero antes de aplicar la técnica 78.
INMUNOBLOT E INMUNOPRECIPITACION
Las dos técnicas detectan antígenos epidérmicos a los que se unen los anticuerpos circulantes de pacientes con pénfigo. La fuente del antígeno en la inmunoprecipitación es el queratinocito cultivado, que se incuba con el suero previamente a la electroforesis en gel. Al no desnaturalizar las proteínas, permite la detección de anticuerpos contra epítopos conformacionales en el pénfigo. La inmunoprecipitación es más sensible que el inmunoblot, pero tiene las desventajas de que requiere trabajar con radiactividad, y es complicada de realizar y cara.
El inmunoblot utiliza extractos de epidermis o dermis para conseguir los antígenos utilizando dodecil sulfato sódico y la electroforesis en gel de poliacrilamida, a los que se añade posteriormente el suero con los autoanticuerpos circulantes. Al producirse una desnaturalización de los antígenos durante el proceso de análisis sólo se detectan anticuerpos que reaccionan contra epítopos secuenciales.
Recientemente se han utilizado ambas técnicas para la detección de la fracción extracelular de las desmogleínas obtenidas de forma recombinante 140.
TRATAMIENTO DEL PÉNFIGO
La tendencia actual en el manejo del pénfigo es individualizar el tratamiento, siendo el objetivo final la supresión total 141-144. Según un estudio la remisión completa se consigue en el 38, 50 y 75 % de los pacientes a los 3, 5 y 10 años, respectivamente, después del diagnóstico 145. Los pacientes con un cuadro clínico moderado o leve y con una respuesta rápida al tratamiento tienen más probabilidades de conseguir una remisión completa. La decisión de suspender el tratamiento se basa en una remisión clínica prolongada y en los hallazgos de la inmunofluorescencia directa (IFD) o indirecta (IFI) 143.
Existe una clara evidencia científica de la efectividad de los corticoides en el tratamiento del pénfigo. Sin embargo, la pauta de administración óptima o la introducción de la terapia adyuvante no está estandarizada. Los estudios controlados del tratamiento del pénfigo son escasos, a menudo se utilizan varios medicamentos, la respuesta al tratamiento no es inmediata, el seguimiento de los pacientes es corto y los tratamientos en muchos casos se establecen sobre la base de las interpretaciones individuales de la literatura médica junto con la experiencia personal de cada autor. La eliminación de los posibles factores desencadenantes, la ingesta de una dieta hipercalórica rica en proteínas y el abordaje de las complicaciones clínicas que aparezcan en la evolución son aspectos importantes que deben tenerse en cuenta.
ESTEROIDES
Corticoides orales
Los esteroides generales han modificado el pronóstico del pénfigo 146. Antes de su utilización, la mortalidad del pénfigo era del 75 %. Cuando se inició el tratamiento con esteroides en la década de 1950 la mortalidad descendió hasta el 30 %. La mortalidad continuó descendiendo en las décadas siguientes hasta un porcentaje del 5,9 % coincidiendo con la introducción de la terapia adyuvante con inmunosupresores. Es dudoso que los inmunosupresores sean los verdaderamente responsables de este descenso, porque la mortalidad disminuyó también de manera drástica en los pacientes tratados únicamente con esteroides. La mejora del pronóstico pudiera ser debida a un diagnóstico y una terapia más temprana de la enfermedad y/o a una mejor utilización de los esteroides orales. En las últimas décadas la mortalidad estimada del pénfigo es inferior al 10 %, la frecuencia se mantiene estacionaria y muchas de estas muertes son de naturaleza iatrogénica 147,148.
El tratamiento más efectivo para el tratamiento del pénfigo son los esteroides orales. Su administración viene condicionada por sus efectos secundarios. Una de las complicaciones más frecuentes a largo plazo del tratamiento prolongado con corticoides es la osteoporosis, que debe manejarse adecuadamente 149. Ello ha impulsado a la búsqueda de un tratamiento adyuvante que permitan disminuir la dosis y el tiempo de administración de esteroides, pero se carece de estudios que demuestren su eficacia con ensayos aleatorizados. El 75 % de los expertos inicia tratamiento con prednisona y, de ellos, el 26 % añade inmunosupresores inmediatamente. El otro 25 % de los expertos inicia el tratamiento con oro, tetraciclina, sulfonas o intenta eliminar los posibles factores desencadenantes como medicamentos, infección concurrente o estrés emocional 150.
Se han diseñado tres estrategias globales para el tratamiento del pénfigo con corticoides orales. La primera fue propuesta por Lever y Schaumburg-Lever, que trataban todos los enfermos con dosis altas y fijas de prednisona 151 (200-400 mg/día durante 6 u 8 semanas), seguido por un rápido descenso a una dosis de mantenimiento de 15 mg/día. La segunda estrategia fue una modificación que realizaron los mismos autores en 1984 152, considerando que podrían existir dos escenarios según la extensión y el curso. Ante un pénfigo que no fuera muy extenso o progresivo se utilizarían dosis intermedias de prednisona en días alternos (40 mg), junto con la administración de un inmunosupresor (azatioprina) al menos durante un año. Ante una enfermedad extensa y progresiva se utilizaría la prednisona a una dosis de 200/400 mg/día durante 5 a 10 semanas para inducir la remisión, y luego la dosis era reducida a 40 mg/día la primera semana, a 30 mg la segunda semana y a 25 mg la tercera semana para continuar luego como las formas no muy extensas. La tercera estrategia fue diseñada por Bystrin 153,154 quien reconoce que dado que la gravedad del pénfigo es variable el esquema debería individualizarse, confiriéndole una flexibilidad acorde con el estado del enfermo, según se muestra en la tabla 4. La finalidad del tratamiento en el pénfigo es inducir una remisión completa que permita suspender el tratamiento y reducir los efectos secundarios a los mínimos posibles. Ello se consigue en tres fases 155:

La primera fase, denominada fase de control, en la cual la intensidad del tratamiento se incrementa rápidamente hasta conseguir suprimir la actividad de la enfermedad. Se debe conseguir una marcada reducción o completa supresión de la aparición de nuevas lesiones, la ausencia de picor y el comienzo de la curación de las lesiones ya existentes. Este periodo debe tener una duración de semanas y no de meses. El pénfigo responde rápidamente a los tratamientos en días en la mayor parte de los casos si la dosis de tratamiento es correcta, y si continúa teniendo actividad clínica es una indicación de tratamiento incorrecto.
La segunda fase es la llamada fase de consolidación, durante la cual la dosis necesaria para el control de la actividad del pénfigo es mantenida hasta que la mayor parte de las lesiones han desaparecido. La longitud de este periodo se mide en semanas, no en meses. Si las lesiones curan lentamente indica que la intensidad del tratamiento es inadecuada y deberá ser aumentada. Las dosis de tratamiento deben ser mantenidas hasta que la mayor parte de las lesiones hayan desaparecido.
La tercera fase, de mantenimiento, ocurre cuando las dosis de tratamiento se descienden paulatinamente hasta conseguir el nivel más bajo de tratamiento que suprime la aparición de nuevas lesiones, con el objetivo de suspender el tratamiento que puede conseguirse en la mayor parte de los enfermos.
Corticoides en terapia pulsátil 156,157
En casos de pénfigo graves o recalcitrantes que no responden a elevadas dosis de esteroides, se utiliza la terapia pulsátil. Su finalidad es conseguir una remisión rápida, minimizando los efectos secundarios de los esteroides, aunque son necesarios más estudios para demostrar la eficacia y la escasez de efectos secundarios. Habitualmente la pauta consiste en la administración intravenosa de dosi s muy altas de esteroides durante un corto periodo de tiempo. Se utiliza la metilprednisona de manera habitual a la dosis de 1 g diario, o bien dexametasona 300 mg, durante 5 días en infusión de 2 o 3 h. Aunque sus resultados son difíciles de evaluar parece inducir remisión en el 50 % de los pacientes en algunos estudios. No obstante, los diversos esquemas utilizados varían ampliamente e incluyen la administración oral del pulso de esteroides, su administración con ciclofosfamida o la administración simultánea de dosis más bajas de esteroides.
Generalmente es bien tolerada en pacientes jóvenes y sanos siendo los efectos secundarios menores. Los más importantes son eritema, trastornos del sueño, cambios de humor y ganancia de peso. No obstante, en enfermos mayores o con enfermedad previa pueden aparecer complicaciones graves, aunque son muy raras. Entre ellos están las convulsiones, hipertensión, trastornos en el balance electrolítico, cardiopatía y pancreatitis.
Corticoides tópicos o intralesionales
El pénfigo puede manifestarse con escasas lesiones o estar localizado durante varios años en una misma zona, y a veces los corticoides tópicos pueden ser suficientes para controlar el cuadro clínico sin los efectos secundarios de los esteroides sistémicos 158,159. En otras ocasiones los corticoides tópicos pueden ayudar a reducir la dosis de esteroides sistémicos 160.
Los corticoides intralesionales 146 pueden ser útiles para tratar un pénfigo de gravedad intermedia que se manifiesta únicamente por escasas lesiones, en lesiones recalcitrantes como aquellas de la mucosa oral y en las nuevas lesiones que aparecen al disminuir la dosis de esteroides. No existe ningún estudio controlado acerca de su eficacia. Y, si después de dos o tres inyecciones en el mismo lugar, la lesión no ha remitido, deberían suspenderse.
TERAPIA ADYUVANTE
Empíricamente con la terapia adyuvante se disminuiría la dosis total de esteroides, pero no existe ningún estudio controlado y prospectivo que compare la dosis acumulativa de esteroides con y sin terapia adyuvante. Bystrin recomienda la terapia adyuvante únicamente si existe contraindicación relativa al uso de esteroides, si aparecen efectos secundarios y si la dosis de esteroides no puede ser reducida por brote de la actividad de la enfermedad. La efectividad de estos agentes no está demostrada en ensayos controlados por lo que la elección de uno u otro estará basada en la experiencia del clínico y el estado del enfermo.
Plasmaféresis 161
Su fundamento es eliminar los autoanticuerpos responsables de la enfermedad, pero muchas veces la plasmaféresis es seguida de un fenómeno de rebote que se explica por la redistribución de los anticuerpos desde el espacio extravascular y/o una síntesis mayor de éstos. Ésta podría ser la razón por la que en estudios controlados y aleatorizados de enfermos tratados con dosis bajas de prednisolona y plamaféresis los resultados no son mejores que con prednisona únicamente 162. Por ello, hoy tiende a utilizarse junto con la administración de inmunosupresores 163,164 con lo que los resultados parecen ser más prometedores. No está claro cuál es el inmunosupresor de elección, aunque algunos se decantan a favor de la ciclofosfamida y otros por el mofetil micofenolato y la azatioprina. Las indicaciones serían en aquellos enfermos que sólo se controlan con dosis inaceptablemente elevadas de esteroides, aquellos que no se controlan o aquellos enfermos en los cuales no es posible descender la dosis de esteroides. Sin embargo, este tratamiento, además de eliminar los autoanticuerpos causa también pérdida de otras inmunoglobulinas, albúmina y factores de la coagulación, hechos que limitan su utilización. Las complicaciones más frecuentes son escalofríos, reacciones alérgicas, fiebre e hipotensión. Muy rara vez aparece edema pulmonar, y diátesis hemorrágica por déficit de plaquetas y de factores de coagulación e infecciones.
Mediante el procedimiento de la inmunoadsorción extracorpórea 165 se extraen distintos componentes del plasma con menores complicaciones que la plasmaféresis. Las ventajas que supondría este procedimiento frente a la plasmaféresis serían una mayor selectividad en la eliminación de los autoanticuerpos patógenos y una reducción en la pérdida de componentes esenciales del plasma y que no precisa reemplazamiento proteico. Se han señalado buenos resultados en el tratamiento del pénfigo, pero es necesario realizar más estudios para confirmar la eficacia y la pauta de tratamiento.
Inmunoglobulinas intravenosas 166-168
A pesar de que no hay estudios controlados su utilización está de actualidad por su eficacia y la relativa ausencia de efectos secundarios. Su eficacia parece confirmarse a medida que se publican nuevos trabajos tanto en el PV 169 como en el PF 170. Se utiliza generalmente a dosis de 2 g/kg/mes dividido en 5 dosis de 0,4 g/kg con la que se obtienen respuestas en el transcurso de semanas permitiendo la reducción de otros tratamientos. La remisión clínica puede ser mantenida con la administración periódica de gammaglobulinas. Su modo de actuación no está aclarado todavía, pero los títulos de autoanticuerpos descienden rápidamente con el tratamiento. Presumiblemente el organismo detecta un exceso de inmunoglobulinas e inicia un proceso catabólico e indiscriminado de éstas, haciendo desaparecer también los autoanticuerpos patogénicos, aunque se han propuesto múltiples mecanismos de acción. Las gammaglobulinas pueden tener otras acciones como bloquear los receptores Fc de las células llamadas reticuloendoteliales. En enfermos cardiacos pueden desencadenar una insuficiencia cardiaca e hipertensión y se han descrito casos de insuficiencia renal. Otros riesgos incluyen la potencial transmisión de otras enfermedades y el coste del tratamiento.
Azatioprina 146,171,172
Es uno de los medicamentos más ampliamente utilizados con terapia adyuvante con la que se han obtenido remisiones entre el 28 y el 45 % de los pacientes. En algún caso se ha utilizado como monoterapia, pero se necesita un periodo de al menos 6 semanas para manifestar su efecto. La dosis utilizadas son alrededor de 1-3 mg/kg/día, aunque hay autores que abogan por dosis mayores, si bien hoy debe individualizarse de acuerdo con los niveles de tiopurina-metil transferasa para evitar la pancitopenia. La azatioprina se debe evitar cuando los niveles son muy bajos, y deberá utilizarse en dosis inferiores (0,5 mg/kg) en aquellos pacientes con niveles enzimáticos muy reducidos. Los efectos secundarios más frecuentes son la pancitopenia, la hepatitis colestática, la toxicidad gonadal y el posible riesgo de neoplasia a largo plazo.
Ciclofosfamida 173-176
La ciclofosfamida puede utilizarse por via oral o en forma de pulsos, asociada o no a esteroides orales, o como tratamiento adyuvante clásico. La ciclofosfamida oral a dosis de 50-200 mg/día (2-2,5 mg/kg/día) es uno de los inmunosupresores más efectivos para el tratamiento del pénfigo. Tiene el inconveniente de los efectos secundarios agudos como pancitopenias, cistitis hemorrágica, o infecciones por Pneumocystis jiroveci, y también a largo plazo como pueden aparecer enfermedades linfoproliferativas, cáncer de vejiga y esterilidad, por lo que precisa estrecha monitorización. La administración de ciclofosfamida en pulsos 177, con corticoides orales (dexametasona 100-136 mg en 3 días consecutivos) seguida de ciclofosfamida oral 50 mg y esteroides en dosis variables es una pauta muy utilizada en la India. La utilización en pulsos tiene la ventaja teórica sobre el tratamiento adyuvante clásico que puede disminuir el riesgo de efectos secundarios y el potencial oncogénico asociado con el uso diario de ciclofosfamida. Es una terapia efectiva con la que se consiguen remisiones duraderas, pero no está exento de complicaciones 178 entre las que destacan eritema, palpitaciones, debilidad generalizada y malestar. Los efectos secundarios son más limitados cuando se utiliza en forma de dosis inmunoablativas 179,180, aunque los resultados de este procedimiento necesitan reevaluación.
Micofenolato 181-183
Utilizado a dosis variables entre 750 mg y 3,5 g/día parece seguro y efectivo como terapia tanto en el PV como en el PF a una dosis estándar entre 35-45 mg/ kg/día. Sus efectos secundarios son dependientes de la dosis incluyendo afectación gastrointestinal y mielosupresión. En el estudio más numeroso de 42 enfermos se obtuvo remisión clínica en el 71 % de pacientes con PV y en el 45 % con PF, definida como ausencia de lesiones durante un mínimo de 4 semanas y una dosis de prednisona menor o igual a 0,15 mg/kg/día. El 77 % de los mismos no tuvo efectos secundarios, y las alteraciones gastrointestinales aparecieron en el 19 %. El coste del tratamiento fue de aproximadamente cuatro veces el de la azatioprina.
Ciclosporina
Aunque inicialmente en algunos casos fue efectiva, un reciente estudio secuencial aleatorizado y controlado que evalúa la eficacia de la prednisolona frente a prednisolona y ciclosporina a la dosis de 5 mg/kg de peso y día concluye que la ciclosporina no añade ventaja al uso de esteroides 184, aun cuando en algún caso puede ser útil 185.
Clorambucilo 186
Es un agente alquilante similar a la ciclofosfamida que afecta preferencialmente a las células B y que tiene mayor poder inmunosupresor que la azatioprina. El clorambucilo carece de la toxicidad vesical de la ciclofosfamida, pero la supresión de la médula ósea y la inducción de leucemia mieloblástica aguda son dos efectos secundarios importantes. En pacientes en los que ha fallado otra terapia inmunosupresora puede ser eficaz.
Metotrexato 187
Inicialmente se ha considerado efectivo, pero su utilización se ha abandonado a causa de sus efectos tóxicos, que ocurren con dosis altas. Se aconseja la utilización del metotrexato en las mismas dosis que se utilizan para el tratamiento de la psoriasis, a las que es efectivo y seguro. No debe utilizarse en aquellos individuos con dosis de esteroides mayores de 60 mg/día por el riesgo de aumentar las infecciones.
Antiinflamatorios
Sales de oro 188
Su interés ha declinado en los últimos años y son pocos los trabajos publicados. En muy pocos casos se ha utilizado como terapia única. Se pauta habitualmente como ahorrador de esteroides de manera semejante a como se utiliza en la artritis reumatoide, con resultados discretos. Se considera ineficaz en el 15-28 % de los casos y efectos colaterales suficientes para interrumpir el tratamiento se observan en el 17-35 % de los enfermos.
Tetraciclinas 189,190
El uso de tetraciclinas con o sin nicotinamida se han utilizado en algunos pacientes, y es eficaz en alguno de ellos, pero los resultados son controvertidos, ya que los estudios no son controlados y el grado de afectación de los pacientes no se especifica. En un estudio la asociación de corticoides y tetraciclina se asoció a una respuesta más rápida, pero los pacientes en el grupo control tenían una respuesta más prolongada 190. Esta terapia adyuvante podría considerarse quizás en casos de pénfigo leves con escasos efectos secundarios.
Sulfona 153
La limitación en el número de casos tratados hace que sea difícil saber si realmente tiene algún papel en el tratamiento del pénfigo. Aunque su administración se asocia a efectos secundarios como pancitopenia, hepatitis, nefrotoxicidad y neuropatía, éstos no son muy frecuentes.
Otros tratamientos
Rituximab (Anti-CD20) 191-195
El rituximab es un anticuerpo monoclonal quimérico múrido humano obtenido por ingeniería genética, que se une al receptor de superficie CD20 de los linfocitos B, induciendo una lisis de los mismos probablemente mediante citotoxicidad celular dependiente de anticuerpo y citotoxicidad dependiente de complemento. Los escasos pacientes con PV y PF tratados sugieren que puede ser una terapia adyuvante válida para los enfermos refractarios a inmunosupresores a dosis de 375 mg/m 2 por vía intravenosa cada semana durante 4 semanas. En ocasiones se produce una mejoría clínica sin disminución de la concentración plasmática de anticuerpos. Los efectos secundarios han sido escasos, pero son necesarios estudios prospectivos que evalúen la eficacia y tolerancia del rituximab.
PÉNFIGO PARANEOPLASICO
Clásicamente se ha definido como una entidad clínica con erosiones mucosas dolorosas y lesiones cutáneas polimorfas localizadas en tronco, extremidades, palmas y plantas asociadas a una neoplasia subyacente, cuyo sustrato histopatológico muestra una degeneración vacuolar junto con necrosis del queratinocito y acantolisis intraepidérmica. La IFD demuestra depósito de IgG y C3 en la superficie de la célula epidérmica y de manera variable a lo largo de la membrana basal. Se demuestran autoanticuerpos circulantes frente a la superficie celular del esófago de mono y sobre el epitelio de la vejiga urinaria de rata y los antígenos responsables son diversos con pesos moleculares que oscilan entre 170 y 250 kDa 8.
Su incidencia es desconocida, pero es menos frecuente que el PV y el PF. En una serie de pacientes con neoplasias la frecuencia fue del 5 %, porcentaje mayor que en los controles, con una edad media al diagnóstico de 64,7 años 196. En el 84 % de los casos el PPN se asociaba con neoplasias hematológicas, las más frecuentes el linfoma no hodgkiniano y leucemia crónica linfocítica, seguidos por el tumor de Castleman asociación frecuente en niños con PPN 197 y el timoma. En el 16 % restante se diagnosticaba una neoplasia no hematológica, de las que más de la mitad eran carcinomas, con frecuencia de páncreas o sarcomas 198.
Manifestaciones clínicas 199-202
La sintomatología es muy polimorfa, siendo el hallazgo más precoz, constante, y a veces único, el desarrollo de lesiones mucosas orales dolorosas que son muy persistentes, incluso después del tratamiento. Estas lesiones consisten en erosiones o ulceraciones mucosas que se entremezclan con necrosis y lesiones liquenoides (fig. 23). Las lesiones son muy extensas y afectan preferencialmente los bordes de la lengua y los labios, mucosa de la boca, y también en algunos pacientes la faringe, laringe, esófago, la conjuntiva y la mucosa genital. Aproximadamente el 30 o 40 % de los pacientes tiene afectación pulmonar, que se manifiesta con disnea progresiva por una bronquiolitis obliterante. El riñón, la vejiga y el músculo estriado y liso pueden estar afectados, y se considera el PPN como un síndrome multiorgánico autoinmune paraneoplásico 203.
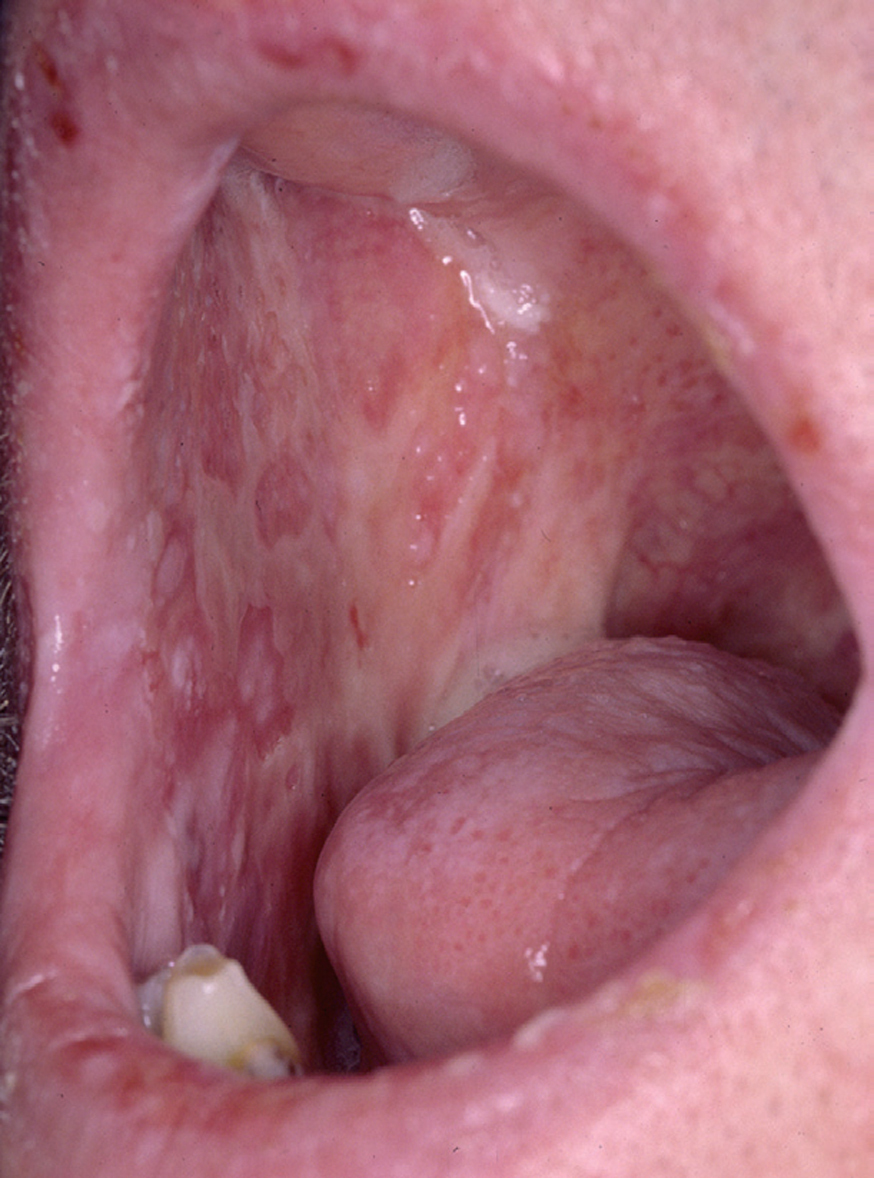
Fig. 23.--Pénfigo paraneoplásico. Cortesía del Dr. L. Requena. Servicio de Dermatología. Fundación Jiménez Díaz.< /FONT>
Las lesiones cutáneas son muy polimorfas y pueden recordar varios cuadros clínicos como el pénfigo, el penfigoide, el eritema exudativo multiforme, el liquen plano y la enfermedad injerto contra huésped (fig. 24). Los diferentes cuadros clínicos pueden coexistir en un mismo paciente o evolucionar de un patrón a otro. Las ampollas aparecen en forma de brotes, y a menudo se rompen y dejan lesiones erosivas en tercio superior de tronco, cabeza y cuello y la parte proximal de las extremidades. En la fase crónica del PPN las lesiones liquenoides pueden predominar sobre las ampollas en la superficie cutánea. En los niños es característico una mucositis oral intensa y lesiones liquenoides cutáneas 197. Las flexuras son uno de los sitios de predilección del PPN. Las lesiones ocasionalmente pueden afectar las palmas y plantas o la región periungueal.
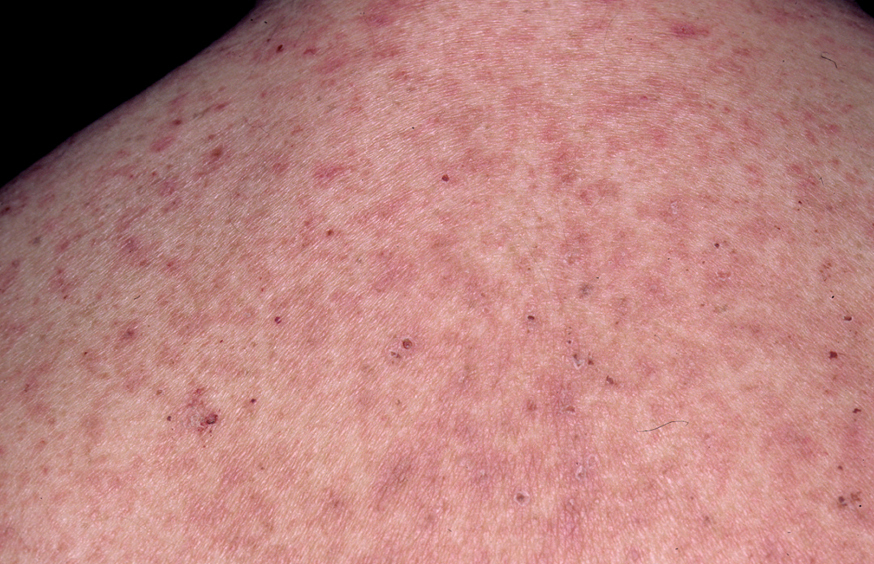
Fig. 24.--Pénfigo paraneoplásico. Cortesía del Dr. L. Requena. Servicio de Dermatología. Fundación Jiménez Díaz.
En contraste con el PV y el PF, en el PPN no se ha observado una buena correlación entre el patrón clínico de enfermedad y el perfil de autoanticuerpos antidesmogleínas 204. La variedad de autoantígenos y la combinación entre ellos condiciona la naturaleza clínica tan polimorfa del PPN.
Histopatología 7,205,206
Los hallazgos histopatológicos son muy variables, en función del tipo de lesión clínica examinada. Las biopsias de las lesiones orales muchas veces muestran una inflamación inespecífica y puede observarse acantolisis suprabasal en el epitelio perilesional. En la piel se puede encontrar desde una ampolla suprabasal con acantolisis hasta una inflamación liquenoide intensa (fig. 25). En la revisión realizada por Horn y Anhalt 205, se encuentran como datos más importantes la acantolisis epidérmica con formación de hendiduras suprabasales, queratinocitos disqueratósicos junto con degeneración vacuolar de los queratinocitos basales y exocitosis epidérmica de células inflamatorias. De acuerdo con ellos la asociación de acantolisis suprabasal y de la necrosis de queratinocitos y/o vacuolización de la capa basal es muy característica del PPN. La asociación de ampollas y acantolisis no tiene nada de peculiar con respecto al PV y es la asociación de la necrosis de los queratinocitos y la ausencia de espongiosis eosinofílica la que sugeriría el cuadro. Las lesiones liquenoides presentan áreas en las que se reconoce un infiltrado linfocitario en dermis superficial con linfocitos individuales infiltrando el epitelio con ocasionales necrosis celulares aisladas.
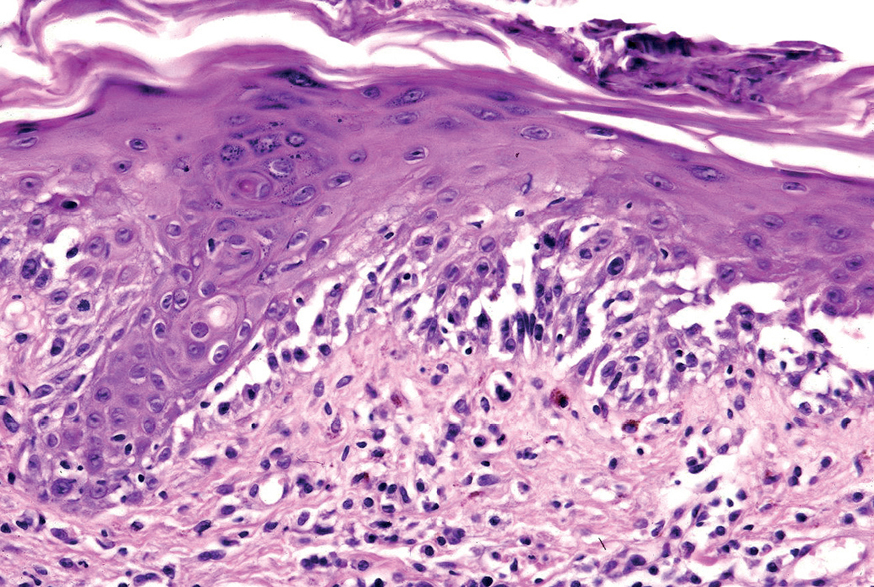
Fig. 25.--Pénfigo paraneoplásico. Cortesía del Dr. L. Requena. Servicio de Dermatología. Fundación Jiménez Díaz.
Inmunopatología
Aunque en algunos casos la IFD muestra falsos negativos, en piel y/o mucosas revela un depósito de IgG con o sin complemento en la superficie celular del queratinocito y/o un depósito lineal granular de IgG con o sin complemento en la membrana basal (fig. 26).
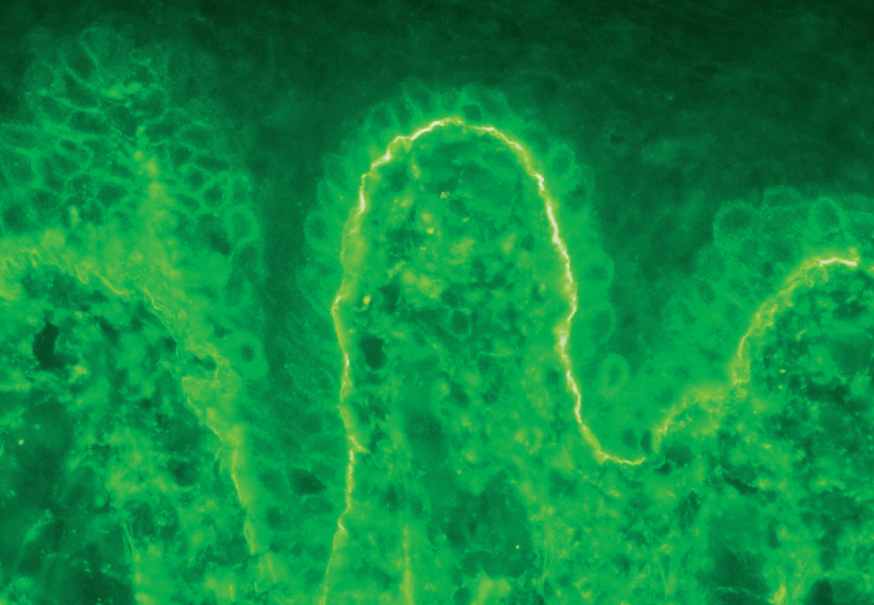
Fig. 26.--Pénfigo paraneoplásico. Cortesía del Dr. L. Requena. Servicio de Dermatología. Fundación Jiménez Díaz.
La IFI demuestra fenómenos más complejos que los observados en el PV o en el PF, poniéndose de manifiesto la presencia de autoanticuerpos circulantes IgG policlonales, con más frecuencia IgG1 e IgG2 que reaccionan con el epitelio del esófago de mono, la vejiga de rata y con otros epitelios como el respiratorio e intestinal. No obstante, el valor de la IFI sobre vejiga de rata ha sido limitado 207. La sensibilidad de la vejiga múrida como sustrato era del 75 %, con una especificidad del 83 %. La IFI en el hígado era específica (96,5 %), pero poco sensible (43 %). La IFI negativa no excluye el diagnóstico de PPN.
Los procedimientos de inmunoblot con extractos celulares epidérmicos, e inmunoprecipitación mediante el uso de extractos de los queratinocitos marcados son más específicos y sensibles para detectar anticuerpos frente a alguna de las proteínas de la familia de las plaquinas. Los autoantígenos indentificados incluyen la desmogleína 3 (130 kDa) y 1 (160 kDa), la envoplaquina (210 kDa), la periplaquina (190 kDa), la desmoplaquina I (250 kDa) y desmoplaquina II (210 kDa), el antígeno 1 del penfigoide (230 kDa), la plectina (400 kDa) y la placoglobina (83 kDa). La frecuencia y la identidad de una banda antigénica de 170 kDa no están bien definidas 200.
Patogenia
La inmunopatología del PPN es más compleja que la del PV y PF. La mayoría de los pacientes con PPN tienen anticuerpos patogénicos contra la desmogleína 3 y 1 135. Es probable que los autoanticuerpos frente a la desmogleína 3 presentes en estos enfermos inicien el proceso de acantolisis y dañen la membrana celular. Una vez dañada se inducirían anticuerpos contra la familia de las plaquinas que están localizadas en el citoplasma de los queratinocitos por un fenómeno de difusión del epítopo 208. Se ha demostrado que la respuesta autoinmune contra la desmogleína 3 en PPN es más amplia que en el PV, ya que se produce frente al dominio amino y carboxiterminal de la proteína, siendo los autoanticuerpos IgG1 y IgG2 las subclases predominantes 209. Las plaquinas son un grupo de proteínas relacionadas secuencialmente que se encuentran en la placa intracelular del desmosoma y hemidesmosoma, siendo una de sus funciones la unión de los filamentos del citoesqueleto a las glucoproteínas de unión transmembranosas. La inmunidad celular parece que está implicada en la citotoxicidad 203, lo que podría explicar el aspecto clínico e histológico de dermatitis liquenoide.
Otra hipótesis sería que proteínas epiteliales tumorales se expresasen constitutivamente o de forma anómala en el tumor, comportándose como antígenos que serían capaces de estimular la respuesta inmunitaria frente a las estructuras epidérmicas. Sin embargo, no existe confirmación de la expresión de proteínas epiteliales en el tumor 200. En algunos casos la producción disregulada de citocinas inducida por las células del tumor estaría implicada en la patogenia, dado que el tratamiento con citocinas puede inducir el PPN 210. La IL-6 se ha encontrado elevada en el PPN, y la IL-6 es capaz de inducir la diferenciación de células B y la producción de inmunoglobulinas 211. Recientemente 212 se ha sugerido que al menos en el PPN asociado a la enfermedad de Castleman, la producción de autoanticuerpos frente a proteínas desmosómicas y hemidesmosómicas es importante en la patogenia de la enfermedad.
Diagnóstico
El diagnóstico del PPN se realiza sobre la base de la presencia de una estomatitis progresiva, dolorosa, con afectación de la lengua con o sin afectación cutánea, una biopsia con acantolisis suprabasal con necrosis de los queratinocitos o dermatitis de interfase o liquenoide, el depósito de autoanticuerpos IgG y complemento en el espacio intercelular epidérmico con o sin depósito en la membrana basal en diversos sustratos, la presencia de inmunoprecipitados contra diferentes proteínas de la familia de las plaquinas y la existencia de una enfermedad linfoproliferativa asociada. Un hallazgo clínico, la asociación con una enfermedad linfoproliferativa y dos hechos biológicos, el marcaje de la vejiga de rata mediante inmunofluorescencia indirecta y el reconocimiento de la banda de envoplaquina y/o periplaquina en el inmunoblot tienen elevada sensibilidad (82-86 %) y especificidad (83-100 %) en el diagnóstico de PPN 213.
Pronóstico
El pronóstico del PPN es malo; el 90 % de los casos fallecen en 2 años. El pronóstico se relaciona con la enfermedad subyacente, la terapia inmunosupresora (sepsis, hemorragia gastrointestinal, fallo multiorgánico) y la insuficiencia respiratoria. El PPN mejora o desaparece en la mayor parte de los individuos con timoma o tumores de Castleman que son tratados quirúrgicamente. Este hecho puede ocurrir 1 o 2 años después de la cirugía. En el caso de tumor de Castleman las lesiones cutáneas mejoraban en 6-11 semanas, las lesiones mucosas mejoraban más tardíamente, y los autoanticuerpos descendían rápidamente 3 semanas después de la extirpación y desaparecían en 5-9 semanas 212.
No hay evidencia de que los regímenes terapéuticos utilizados sean efectivos. Se han utilizado los esteroides en dosis de 0,5-1 mg/kg/día con una respuesta mejor en las lesiones cutáneas que en las orales, habiéndose utilizado la ciclofosfamida, azatioprina, oro, sulfona, plasmaféresis, fotoféresis e inmunoglobulina IV. Algunos pacientes con linfoma no hodgkiniano folicular han mejorado con rituximab 214. La afectación respiratoria parece ser una importante causa de muerte con una enfermedad obstructiva que desemboca en una bronquiolitis obliterante, quizá debida a los autoanticuerpos antidesmoplaquinas presentes en el epitelio respiratorio 215,200.
PÉNFIGO HERPETIFORME
El PH es una entidad que se caracteriza por unas manifestaciones clínicas que recuerdan a la dermatitis herpetiforme, incluyendo la morfología clínica, el prurito y la respuesta terapéutica a sulfonas, y por hechos histopatológicos e inmunopatológicos del pénfigo. Epidemiológicamente es una enfermedad poco frecuente. Se observa en alrededor del 7 % de todos los pénfigos en la serie de Jablonska 216. La edad de aparición es variable, siendo más frecuente entre los 30 y 80 años y afecta a ambos sexos por igual. Clínicamente se manifiesta como una erupción de lesiones cutáneas eritematosas, urticarianas, eritematovesiculosas, papulosas o ampollosas, con lesiones coalescentes de morfología anular, con frecuencia con una distribución herpetiforme, y habitualmente pruriginosas. La afectación mucosa puede ocurrir en alrededor de un tercio de los casos. Clínicamente el cuadro es atípico y puede remedar muchas enfermedades ampollosas incluyendo penfigoide, dermatitis herpetiforme, dermatosis IgA lineal y en ocasiones PF o fogo selvagem217 (fig. 27). Se han descrito casos aislados asociados a tumores 218,219, lupus eritematoso 220, psoriasis 221 o tras la radiación UV 222.
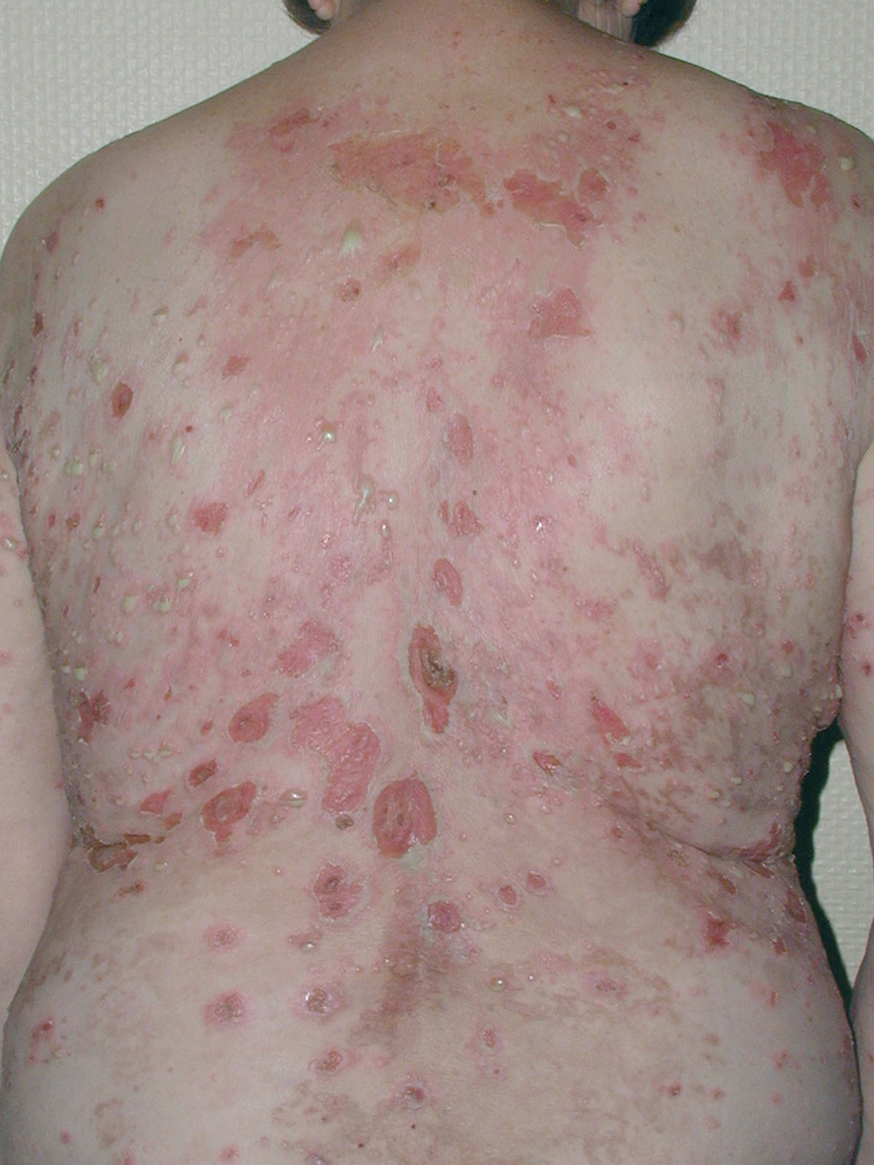
Fig. 27.--Ampollas y erosiones en la espalda delimitadas por pápulas, vesículas y pústulas. Cortesía de Arranz D. et al. Pénfigo herpetiforme asociado a carcinoma del esófago. Actas Dermosifiliogr 2005;96:119-121.
La histopatología es muy polimorfa, con hallazgos que varían en función del tipo de lesión cutánea y de la evolución del cuadro clínico 3. La presencia de vesículas intraepidérmicas, o ampollas subcórneas 222 que están llenas de neutrófilos y eosinófilos en proporción variable es un hallazgo característico (fig. 28). En algunas ocasiones se precisan varias biopsias para llegar a un diagnóstico correcto 6,220, con discreta acantolisis en las capas superiores de la epidermis y espongiosis eosinofílica.
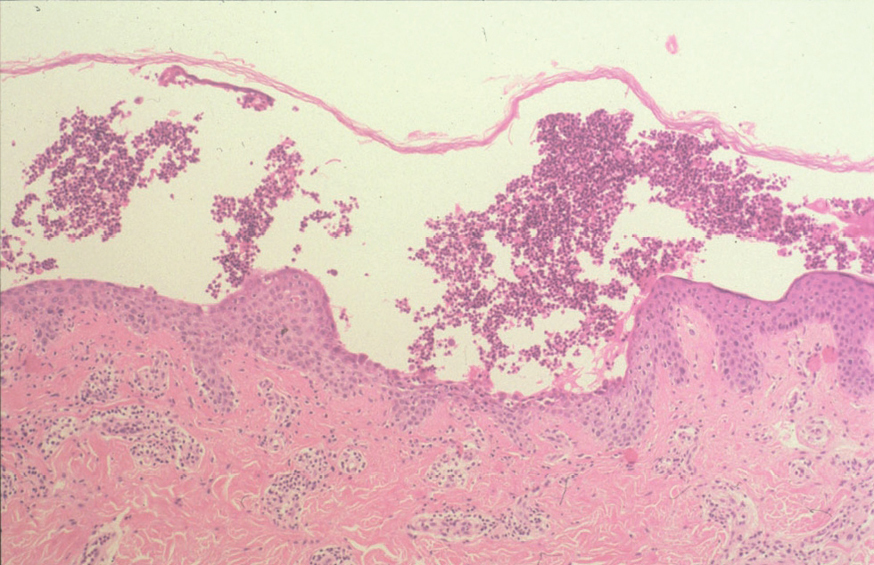
Fig. 28.--Pústula intraepidérmica con eosinófilos y acantolisis. (Hematoxilina-eosina, x20.) Cortesía de Arranz D. et al. Pénfigo herpetiforme asociado a carcinoma del esófago. Actas Dermosifiliogr 2005;96:119-121.
La inmunopatología es importante para confirmar el diagnóstico. La IFD de la piel perilesional muestra la presencia de depósitos IgG y C3 en la superficie del queratinocito en la epidermis superior o en toda la epidermis. La IFI demuestra la presencia de IgG, singularmente IgG4 circulante frente a la superficie celular del queratinocito. Exhibe algunas características clínicas e histopatológicas comunes con el pénfigo IgA del que puede ser diferenciado porque los autoanticuerpos son de la clase IgG. No obstante, se ha descrito recientemente un caso de un PH con anticuerpos IgA e IgG frente a la desmogleína 1 e IgG frente a la desmocolina 223.
Los antígenos diana del PH han sido motivo de discusión, pero hoy se considera que los anticuerpos más frecuentemente se dirigen contra la desmogleína 1. Al encontrarse localizada fundamentalmente en la epidermis superior explicaría la patología preferencial en las capas superiores de la epidermis, y los hallazgos tanto clínicos como patológicos son semejantes al PF 223. No obstante, se han descrito casos en los cuales la reactividad es únicamente frente a la desmogleína 3 224. En otros casos los antígenos pueden ser múltiples 225. El estudio de series de pacientes muestran que el antígeno más frecuente es la desmogleína 1 y en algunos casos puede encontrarse la desmogleína 3 4.
La presentación clínica del PH tan diferente a la del PV, así como la escasez de la acantolisis a pesar de que el sustrato antigénico es el del PV, se han tratado de explicar porque los autoanticuerpos reconocen diferentes epítopos en las moléculas antigénicas. Se ha demostrado por inmunohistoquímica una intensa expresión de IL-8, colocalizada con la IgG en la epidermis superior, precisamente donde ocurre la acantolisis. Los queratinocitos son capaces de segregar mucha mayor cantidad de IL-8 que los controles, de manera que los autoanticuerpos IgG se ligarían a la desmogleína 1 del queratinocito al que inducirían a producir IL-8. Esta citocina sería quimioatractiva para los neutrófilos, que una vez en el foco inflamatorio liberarían proteasas y darían lugar a la formación de ampollas 226.
El tratamiento con sulfona y pequeñas dosis de esteroides ofrece buenos resultados, aun cuando en algunos casos es necesario utilizar inmunosupresores. La sulfona es el medicamento de primera elección en dosis de 100 a 300 mg/día, a la que responden mejor aquellos con bajas concentraciones de autoanticuerpos o los que tienen espongiosis eosinofílica. En la serie de Maciejowska 216 sólo un caso de 15 respondía al tratamiento único con sulfona. Los esteroides sistémicos se requieren muchas veces para conseguir la remisión clínica, que se alcanza aproximadamente a la mitad en la serie citada. En el resto de los pacientes se han utilizado también inmunosupresores como la azatioprina y la ciclofosfamida, y plasmaféresis en algún caso.
PÉNFIGO IgA
Recientemente se ha descrito un grupo poco frecuente de enfermedades autoinmunes ampollosas caracterizadas clínicamente por una erupción vesiculopustulosa, con una histología que muestra infiltración neutrofílica con acantolisis, una IFD con depósito IgA intercelular intraepidérmica y una IFI con presencia de autoanticuerpos circulantes IgA frente a componentes celulares de la epidermis.
La sintomatología e histopatología del pénfigo IgA se ha agrupado en dos patrones: el patrón tipo dermatosis pustulosa subcórnea (DPS) y el tipo dermatosis neutrofílica intraepidérmica (DNI) 227-232. La morfología clínica del tipo DPS, el más frecuente, puede ser indistinguible de la dermatosis pustulosa subcórnea clásica, que consiste en una erupción vesiculopustulosa superficial sobre piel de aspecto normal o eritematosa, con predilección en áreas intertriginosas 233. El signo de Nikolsky es negativo, y las mucosas no están afectadas por lo general. En estos pacientes es conveniente descartar un mieloma IgA o una gammapatía monoclonal IgA que se asocia en el 20 % de los casos a la variante tipo DPS. Las lesiones tipo DNI consisten en lesiones pustulosas que confluyen en placas que adoptan un patrón anular o circinado centradas por costras y pruriginosas. Las lesiones predominan en las axilas e ingles, tronco y en la región proximal de las extremidades 234, y es muy rara la afectación de las mucosas (figs. 29 y 30). Aparece a una edad media o avanzada, con discreto predominio en mujeres. Sin embargo, hay casos en los que la descripción clínica no permite encuadrarlos en cualquiera de los tipos anteriores 235.
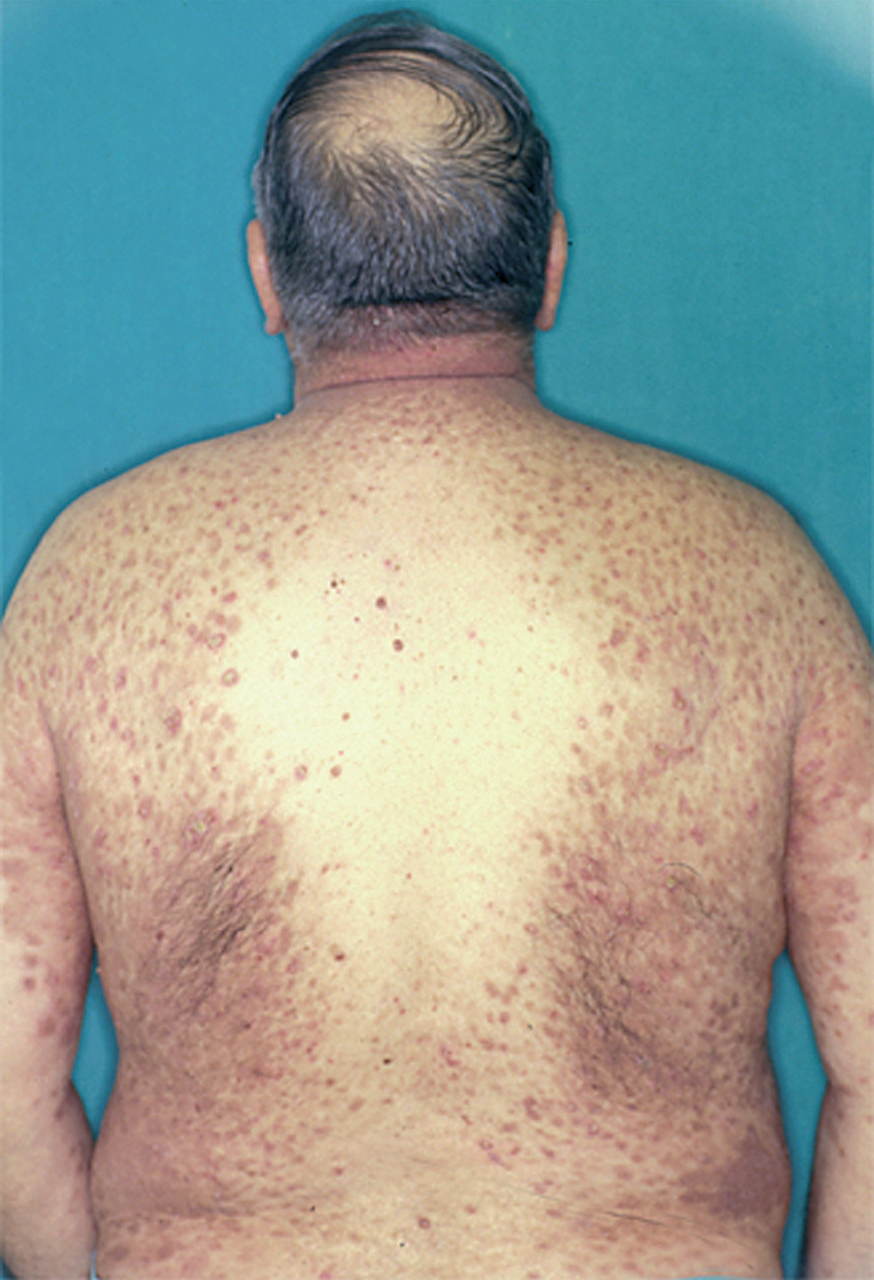
Fig. 29.--Pénfigo IgA tipo dermatosis neutrofílica intraepidérmica.
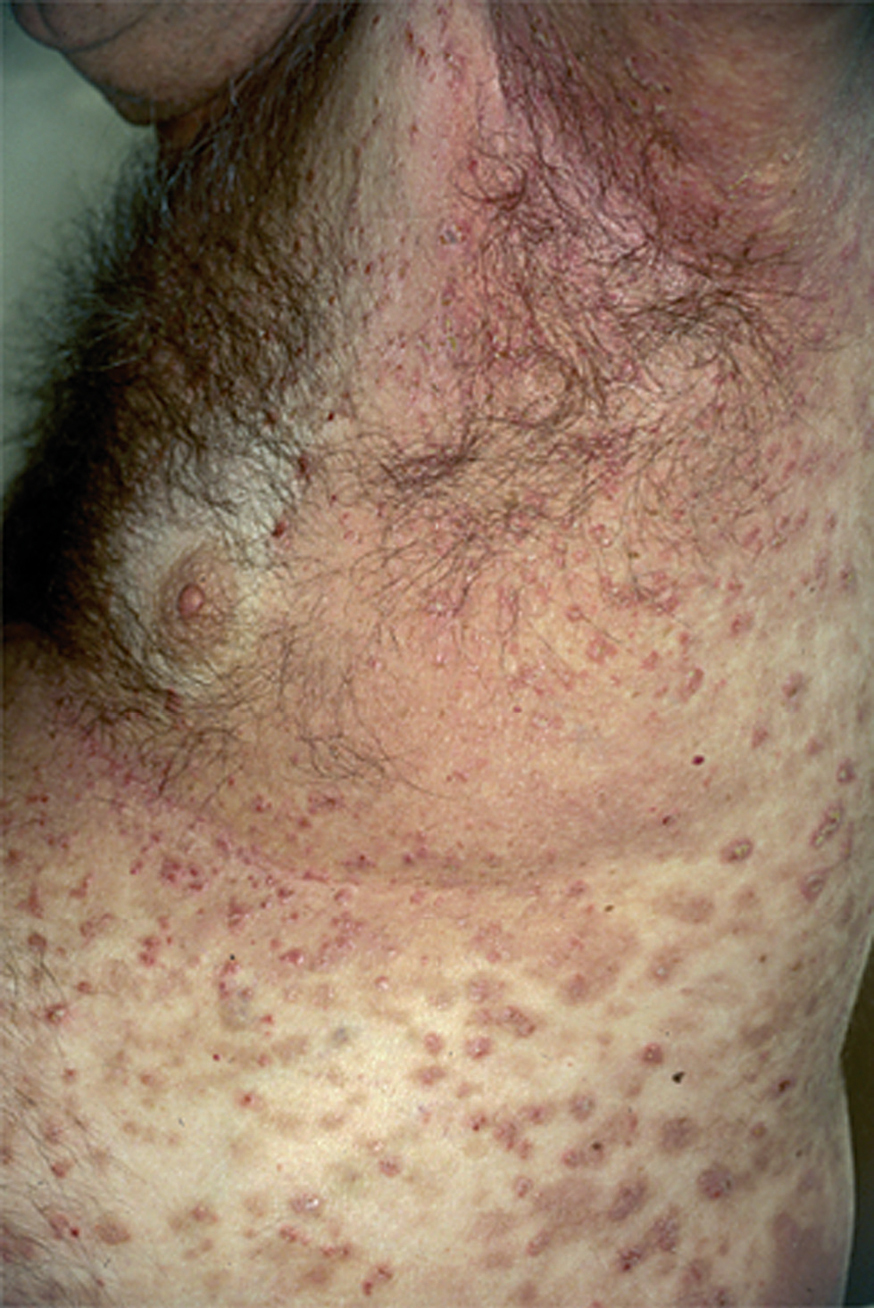
Fig. 30.--Pénfigo IgA tipo dermatosis neutrofílica intraepidérmica.
Histológicamente el tipo DPS muestra vesículas y/o pústulas subcórneas, infiltración neutrofílica con o sin eosinófilos y escasa acantolisis. El tipo DNI se caracteriza por vesículas y/o pústulas suprabasales, infiltración neutrofílica y escasa acantolisis. En la dermis se observa en los dos patrones clínicos un infiltrado perivascular de neutrófilos, eosinófilos y linfocitos.
En la inmunofluorescencia directa en piel perilesional se observa depósito de IgA intercelular con un patrón similar al PV siendo la prueba diagnóstica en estos pacientes. Se observan depósitos subcórneos en el tipo DPS, y suprabasales o en toda la epidermis en el tipo DNI. La IFI en piel humana normal, esófago de mono y cerdo de guinea es positiva en alrededor del 50 % de los casos con autoanticuerpos circulantes del subtipo IgA1229,236, a concentraciones bajas frente a la superficie celular de la epidermis 237.
Mediante la transfección de la desmocolina a las células COS7 derivadas del riñón de mono se ha demostrado que la desmocolina 1 es el antígeno frente al cual van dirigidos los anticuerpos en el tipo DPS 235. Sin embargo, aún no es posible la detección de autoanticuerpos IgA mediante la técnica de ELISA utilizando como antígeno la desmocolina 1, ni por inmunoblot ni por inmunoprecipitación 238. El antígeno diana de la DNI es desconocido, habiéndose propuesto una proteína transmembrana no desmosómica 239. Se desconoce el mecanismo de formación de las lesiones en el pénfigo IgA, pero se cree que la unión del anticuerpo con la desmocolina u otro autoantígeno activaría una respuesta inflamatoria, con un intenso infiltrado neutrofílico, que condicionaría la aparición de lesiones pustulosas.
La sulfona es el tratamiento de primera elección, habitualmente a dosis de 100 mg/día asociado o no a corticoides orales y tópicos 5,6. Si el tratamiento no es eficaz o no se tolera se puede utilizar acitretino 240 a dosis de 20-30 mg/día con o sin PUVA. La combinación de sulfona y acitretino puede ser eficaz en algún caso, y otras veces la colchicina 6,241.



