


Las displasias ectodérmicas son un amplio grupo de trastornos hereditarios que se caracterizan por la alteración de estructuras derivadas del ectodermo. Aunque algunos de estos síndromes poseen características específicas, determinados rasgos clínicos son comunes en muchos de ellos. De modo general, se diferencian 2 grupos de trastornos: uno caracterizado por la aplasia o hipoplasia de los derivados ectodérmicos, que fracasan en su desarrollo y diferenciación por la ausencia de señales recíprocas específicas entre ectodermo y mesénquima, y otro en el que la característica más llamativa es la queratodermia palmoplantar, que se presenta en asociación con otras manifestaciones cuando se afectan otros epitelios altamente especializados. En las últimas décadas se ha logrado identificar el gen responsable en al menos 30 entidades, permitiéndonos entender los mecanismos patogénicos y su correlación con la clínica.
The ectodermal dysplasias are a large group of hereditary disorders characterized by alterations of structures of ectodermal origin. Although some syndromes can have specific features, many of them share common clinical characteristics. Two main groups of ectodermal dysplasias can be distinguished. One group is characterized by aplasia or hypoplasia of ectodermal tissues, which fail to develop and differentiate because of a lack of reciprocal signaling between ectoderm and mesoderm, the other has palmoplantar keratoderma as its most striking feature, with additional manifestations when other highly specialized epithelia are also involved. In recent decades, the genes responsible for at least 30 different types of ectodermal dysplasia have been identified, throwing light on the pathogenic mechanisms involved and their correlation with clinical findings.
El ectodermo es uno de los componentes embrionarios primordiales. En torno a la tercera semana de desarrollo experimenta una subdivisión que dará lugar al neuroectodermo, origen del sistema nervioso, y al ectodermo, que recubrirá toda la superficie embrionaria y formará la epidermis, sus anexos y el esmalte dental. Por tanto, del ectodermo derivan no solo el pelo, los dientes, las uñas y las glándulas sudoríparas, sino también el sistema nervioso central, el sistema nervioso periférico, el ojo, el oído y la nariz, así como las glándulas ecrinas, mamarias y pituitarias1. Por otro lado, el ectodermo mantiene complejas interacciones con el mesodermo durante su desarrollo, lo que explica que su alteración conlleve, en algunos casos, anomalías asociadas en estructuras mesodérmicas como los sistemas musculoesquelético y genitourinario2.
Las displasias ectodérmicas (DE) son un grupo heterogéneo de trastornos hereditarios caracterizados por compartir anomalías estructurales y funcionales en varios tejidos derivados del ectodermo. La mayoría de estas enfermedades asocian también un desarrollo anormal de estructuras derivadas del mesodermo y, ocasionalmente, retraso mental. Son consideradas enfermedades raras, con una incidencia estimada de 7/10.000 nacimientos3. Pueden seguir cualquiera de los modelos posibles de herencia mendeliana4, y aunque las características clínicas son comunes en muchas de ellas, algunos síndromes poseen hallazgos clínicos específicos. Hasta la fecha se conocen unas 200 entidades diferentes, de las cuales se ha identificado el gen causal en aproximadamente 30. Las mutaciones en tan solo 4 genes (EDA1, EDAR, EDARADD y WNT10A) son responsables de la mayoría de los casos de DE5.
Recuerdo históricoLas primeras descripciones de casos clínicos que podrían corresponderse con lo que ahora conocemos como DE se remontan a 17926. En 1848 Thurman definió la displasia ectodérmica anhidrótica (DEH) como una entidad independiente7. Posteriormente se describieron casos similares como el presentado por Wedderhorn y publicado en 1875 por el naturalista Charles Darwin: «I may give an analogous case, communicated to me by Mr. W. Weddenburn, of a Hindoo family in Scinde, in which ten men, in the course of four generations, were furnished, in both jaws taken together, with only four small and weak incisor teeth and with eight posterior molars. The men thus affected have very little hair on the body, and become bald early in life. The also suffer much during hot weather from excesive dryness of the skin. It is remarkable that no instance has ocurred of a daughter being affected… though the daughters in the above family are never affected, the transmit the tendency to their sons: and no case has occurred of a son transmitting it to his sons. The affection thus appears only in alternate generation, or after long intervals»8. El caso que aquí nos describía Darwin se correspondería con lo que hoy conocemos por displasia ectodérmica anhidrótica ligada al X (DEH-X), término que finalmente fue acuñado por Weech en 19299.
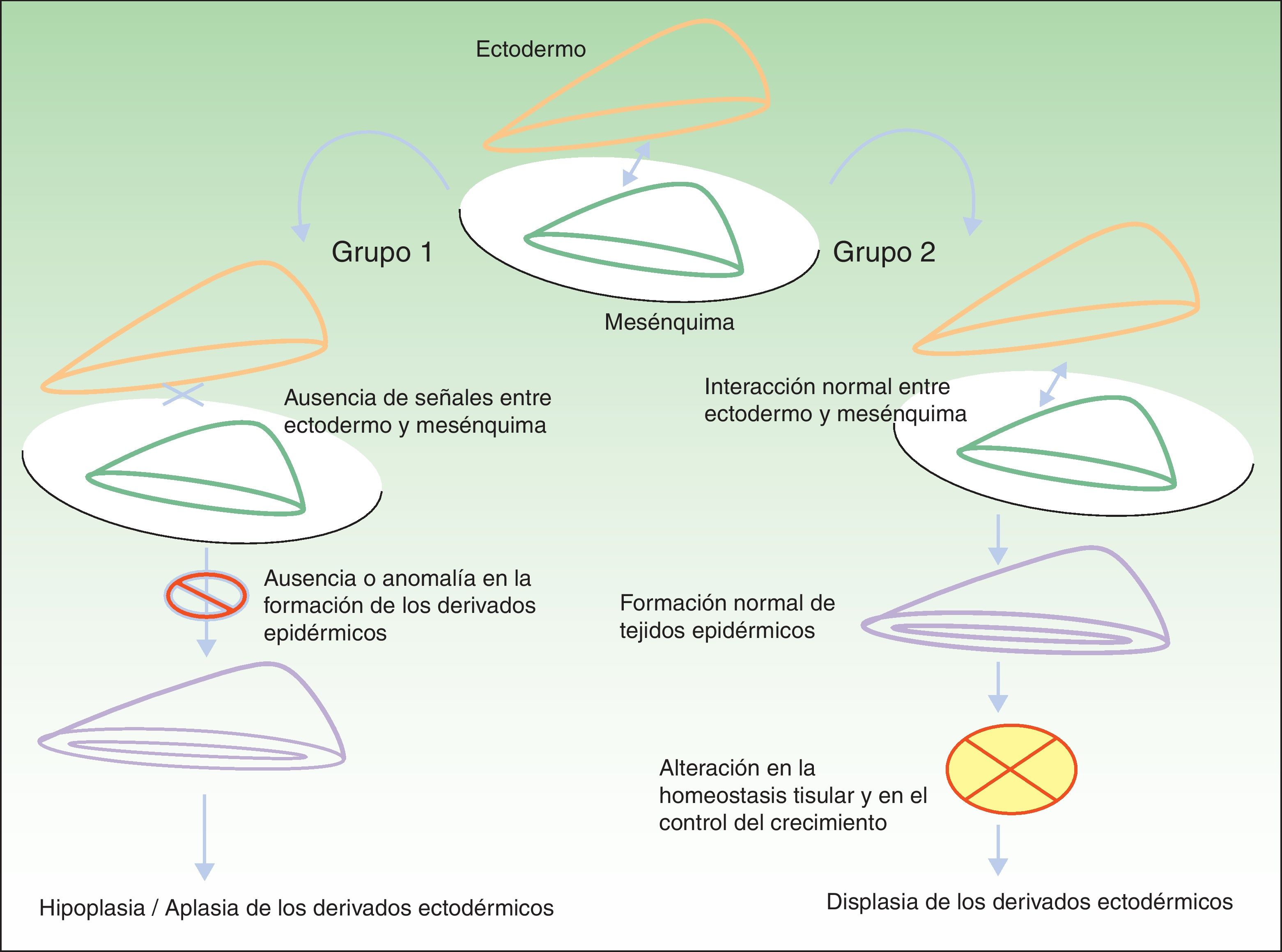
Clasificación de las displasias ectodérmicasLa categorización de las DE es compleja, por lo que los sistemas de clasificación se han ido sucediendo con el objeto de compaginar datos clínicos y genéticos10–15. Los hallazgos biomoleculares han permitido saber que los genes causales actúan básicamente a través de 2 mecanismos patogénicos, los cuales se corresponden con unos hallazgos clínicos concretos para cada caso. Basándose en los mismos, Priolo estableció en 2009 una clasificación clínico-funcional2, que es la que vamos a seguir en esta revisión. Este autor propuso diferenciar 2 grupos de trastornos (fig. 1):

Mecanismos patogénicos en las displasias ectodérmicas. En el grupo 1 se altera la interacción entre el ectodermo y el mesénquima, lo cual impide la correcta diferenciación de los derivados epidérmicos, que son hipoplásicos o aplásicos. En el grupo 2 la interacción entre el ectodermo y el mesénquima es normal, y los derivados epidérmicos se diferencian con normalidad, pero la homeostasis tisular y el crecimiento son anómalos, por lo que existe displasia de los derivados ectodérmicos. Modificada de Priolo M2.
Trastornos en los que puede reconocerse un defecto en la interacción entre ectodermo y mesénquima. Se han identificado 2 mecanismos fisiopatológicos:
- 1.
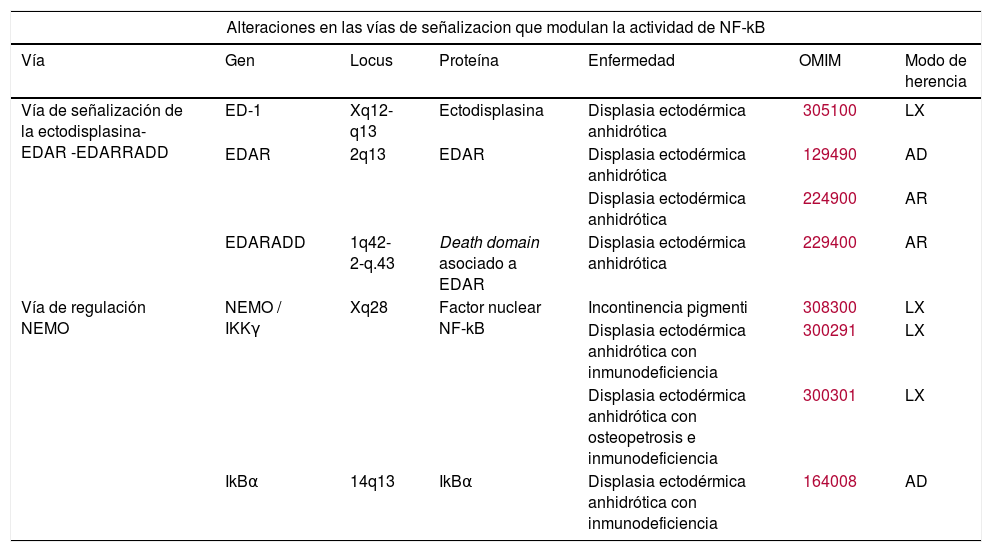
Alteración en las vías de señalización que modulan la actividad de la molécula factor nuclear kappa beta (NF-kB) (vía de señalización ectodisplasina/EDAR/EDARADD y vía de regulación NEMO).
- 2.
Alteración de reguladores de la transcripción y/o de la expresión de genes, como p63, DLX3, MSX1, EVC2 y EVC.
Clínicamente, el fenotipo resultante es una hipo/aplasia de los derivados ectodérmicos, que fracasan en su desarrollo y diferenciación por la ausencia de señales recíprocas específicas entre ectodermo y mesénquima (tabla 1).
Trastornos del grupo 1
| Alteraciones en las vías de señalizacion que modulan la actividad de NF-kB | ||||||
|---|---|---|---|---|---|---|
| Vía | Gen | Locus | Proteína | Enfermedad | OMIM | Modo de herencia |
| Vía de señalización de la ectodisplasina- EDAR -EDARRADD | ED-1 | Xq12-q13 | Ectodisplasina | Displasia ectodérmica anhidrótica | 305100 | LX |
| EDAR | 2q13 | EDAR | Displasia ectodérmica anhidrótica | 129490 | AD | |
| Displasia ectodérmica anhidrótica | 224900 | AR | ||||
| EDARADD | 1q42-2-q.43 | Death domain asociado a EDAR | Displasia ectodérmica anhidrótica | 229400 | AR | |
| Vía de regulación NEMO | NEMO / IKKγ | Xq28 | Factor nuclear NF-kB | Incontinencia pigmenti | 308300 | LX |
| Displasia ectodérmica anhidrótica con inmunodeficiencia | 300291 | LX | ||||
| Displasia ectodérmica anhidrótica con osteopetrosis e inmunodeficiencia | 300301 | LX | ||||
| IkBα | 14q13 | IkBα | Displasia ectodérmica anhidrótica con inmunodeficiencia | 164008 | AD | |
| Alteración de reguladores de la transcripción/expresión de genes | |||||
|---|---|---|---|---|---|
| Gen | Locus | Proteína | Enfermedad | OMIM | Modo de herencia |
| p63 | 3q27 | P63 | Síndrome EEC | 604292 | AD |
| Síndrome AEC | 106260 | AD | |||
| Síndrome ADULT | 103285 | AD | |||
| Síndrome limb-mammary | 603543 | AD | |||
| Síndrome de Rapp-Hodgkin | 603543 | AD | |||
| DLX3 | 17q21 | DLX3 | Síndrome trico-dento-óseo | 190320 | AD |
| MSX1 | 4p16.1 | MSX1 | Enfermedad de Witkop | 189500 | AD |
| EVC2 | 4p16 | EVC2 | Síndrome Ellis-Van-Creveld | 225500 | AR |
| EVC | 4p16 | EVC | Disostosis acrodental de Weyers | 193530 | AD |
| Síndrome Ellis-Van-Creveld | 225500 | AR | |||
Modificada de Priolo M2. AD: herencia autosómica dominante; AR: herencia autosómica recesiva; LX: herencia ligada a X; OMIM: Online Mendelian Inheritance in Man.
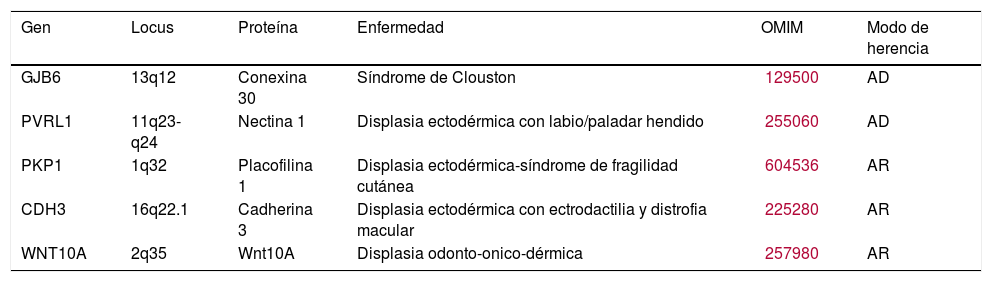
Trastornos en los que existe una función anormal de una proteína estructural localizada en la membrana celular, como la nectina 1, las conexinas o la placofilina, cuyo papel en la adhesión y comunicación intercelular es crucial para el mantenimiento de la homeostasis tisular, el control del crecimiento, el desarrollo y la respuesta celular sincronizada a diferentes estímulos.
Clínicamente, estos trastornos se caracterizan principalmente por anomalías dermatológicas como la queratodermia palmoplantar, con o sin afectación de epitelios altamente diferenciados, que se traducen en sordera o distrofia de la retina (tabla 2).
Trastornos del grupo 2
| Gen | Locus | Proteína | Enfermedad | OMIM | Modo de herencia |
|---|---|---|---|---|---|
| GJB6 | 13q12 | Conexina 30 | Síndrome de Clouston | 129500 | AD |
| PVRL1 | 11q23-q24 | Nectina 1 | Displasia ectodérmica con labio/paladar hendido | 255060 | AD |
| PKP1 | 1q32 | Placofilina 1 | Displasia ectodérmica-síndrome de fragilidad cutánea | 604536 | AR |
| CDH3 | 16q22.1 | Cadherina 3 | Displasia ectodérmica con ectrodactilia y distrofia macular | 225280 | AR |
| WNT10A | 2q35 | Wnt10A | Displasia odonto-onico-dérmica | 257980 | AR |
Modificada de Priolo M2. AD: herencia autosómica dominante; AR: herencia autosómica recesiva; OMIM: Online Mendelian Inheritance in Man.
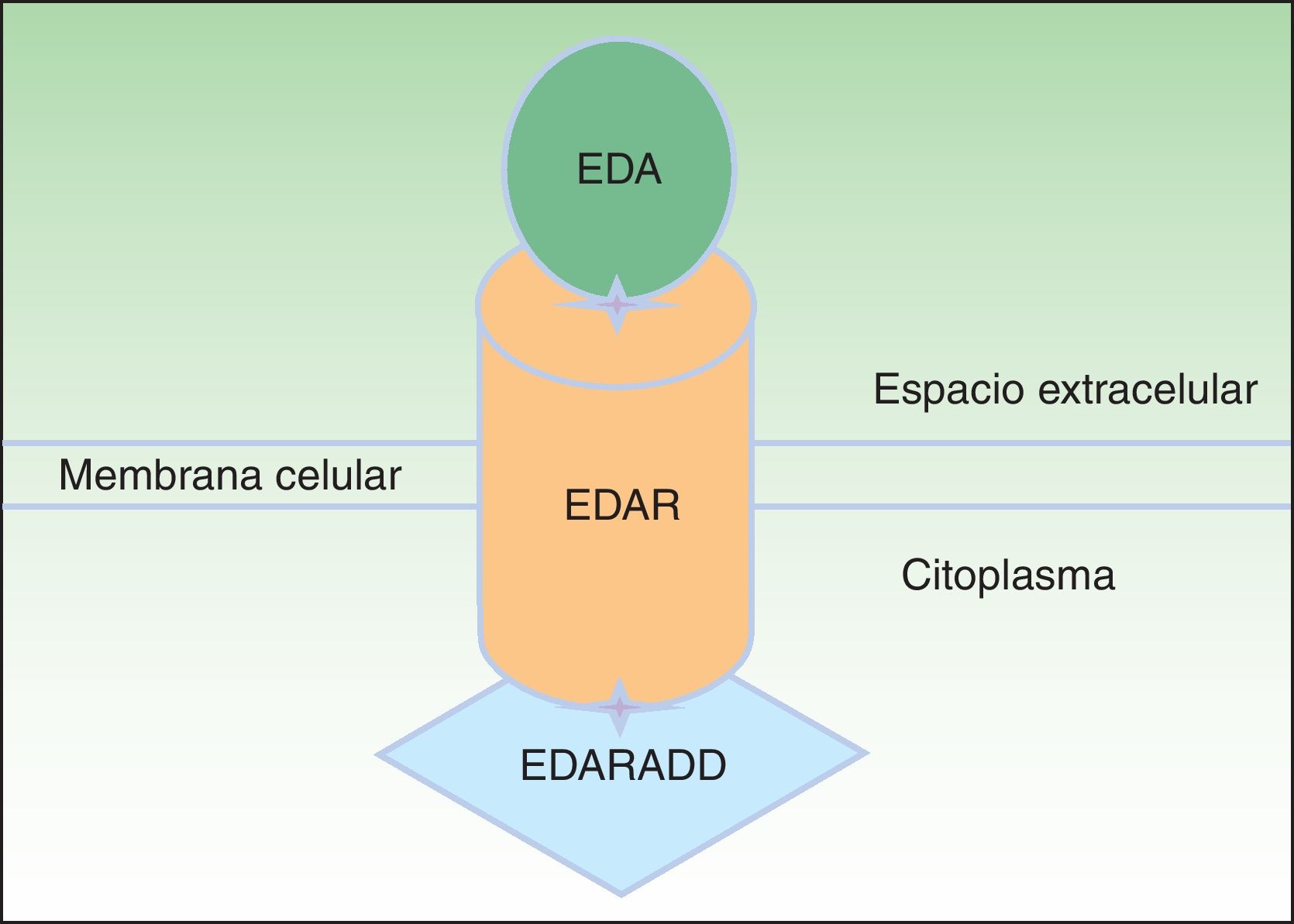
La proteína ectodisplasina-A (EDA), constituida por 391 aminoácidos, es una proteína transmembrana tipo ii que pertenece a la familia del factor de necrosis tumoral (TNF)16. Entre las distintas isoformas de EDA, las biológicamente más relevantes son la EDA-A1 y la EDA-A217. La EDA-A1 se une al receptor de la EDA (EDAR), mientras que la EDA-A2 se une al EDAR A2 ligada al X (XEDAR)18 (fig. 2). La EDAR es una proteína transmembrana tipo i, que consta de 448 aminoácidos, y pertenece a la superfamilia de los receptores del TNF (TNFR). Contiene una región extracelular, una región transmembrana y un death domain en su región intracelular19. El death domain es un módulo de interacción proteico que interacciona con los death domains de otras proteínas activando cascadas metabólicas que con frecuencia están implicadas en la regulación de la apoptosis y la inflamación a través de la cascada NFkB. El dominio extracelular de la EDAR es esencial para ligarse a EDA-A1, mientras que el death domain de la región intracelular desempeña un papel importante para el inicio de las señales de transducción que conducen a la apoptosis20. El death domain de EDAR se adapta al death domain de la EDARADD, una proteína de 208 aminoácidos21, por lo que la integridad de los death domain de ambas proteínas es vital para su interacción22 y la normal regulación de la morfogénesis embrionaria17. Por tanto, la vía de señalización mediada por la EDA es fundamental para el desarrollo apropiado de varios órganos y estructuras derivadas del ectodermo, como el pelo, las uñas, la glándula pituitaria, las glándulas mamarias y sudoríparas, la nariz, los ojos y el esmalte dentario23.
 se une al receptor de la ectodisplasina-A (EDAR), localizado en la región extracelular de EDAR. EDAR contiene además una región transmembrana y un death domain en la región intracelular, a través del cual se une con el death domain de EDARADD. Modificada de Lu PD et al.36.")
Vía de la ectodisplasina-EDAR-EDARADD. La proteína ectodisplasina-A (EDA) se une al receptor de la ectodisplasina-A (EDAR), localizado en la región extracelular de EDAR. EDAR contiene además una región transmembrana y un death domain en la región intracelular, a través del cual se une con el death domain de EDARADD. Modificada de Lu PD et al.36.
El gen EDAR, de 12 exones, se localiza en el cromosoma 2 (locus 2q11-q13)19 y codifica al receptor para EDA-A1 (EDAR)17. Hasta la fecha se han descrito al menos 41 mutaciones en el gen EDAR, la mayoría de las cuales afectan al exón 12, que codifica la región C-terminal donde se encuentra el death domain24. Algunas de estas mutaciones han sido descritas en pacientes españoles25,26. El gen EDARADD, localizado en el cromosoma 1 (locus 1q-42-q43), codifica la EDARADD, y está implicado en cuadros autosómicos tanto recesivos como dominantes17,27. Por último, recientemente se ha demostrado que algunas mutaciones en el gen WNT10A, un miembro de la vía de señalización Wnt, implicada en el desarrollo embriogénico y la diferenciación celular, así como en ciertos procesos fisiológicos del adulto y en el cáncer, dan lugar a varias formas de DE con herencia autosómica, como la DEH o la displasia onico-odonto-dérmica5.
Displasia ectodérmica anhidrótica ligada al cromosoma XTambién conocida como síndrome de Christ-Siemens-Touraine, se trata de la forma de DE más frecuente, con una incidencia aproximada de 1 caso por cada 100.000 nacimientos28. Por el modo de herencia ligada a X, los hombres afectados muestran todas las características típicas de la enfermedad o gran parte de ellas, mientras que las mujeres portadoras solo muestran manifestaciones parciales. No existe relación geno-fenotípica, y hay una gran variación en el fenotipo tanto entre distintas familias como dentro del mismo grupo familiar29. La DE anhidrótica ligada al cromosoma X (DEH-X) se produce como resultado de alteraciones en el gen ED1, también denominado EDA30. Este gen se localiza en el brazo largo del cromosoma X (locus Xq12-q13) y codifica la proteína EDA. Se han identificado más de 204 mutaciones diferentes en el gen ED124. Aunque se han descrito extensas deleciones e inserciones, el 80% de los casos presenta una mutación31,32. Recientemente, un grupo español ha identificado una familia con una mutación previamente no descrita en este gen33.
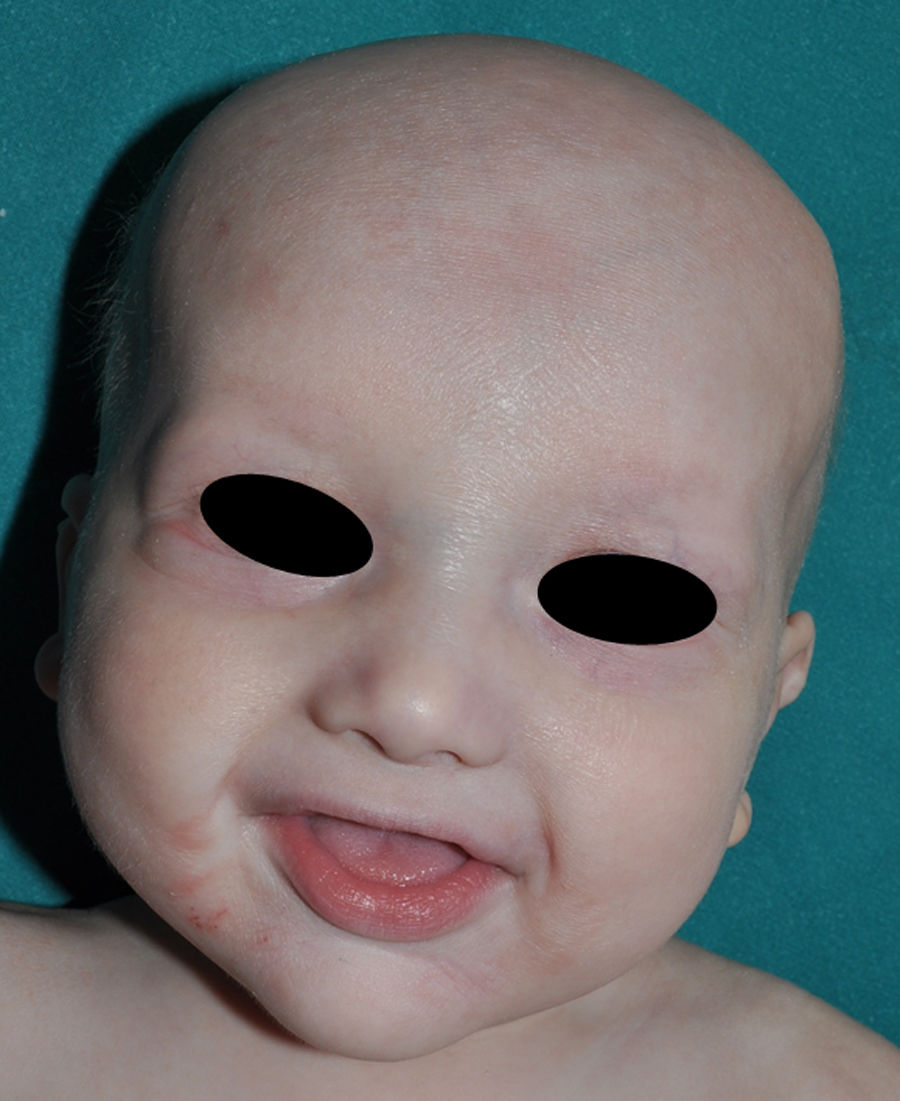
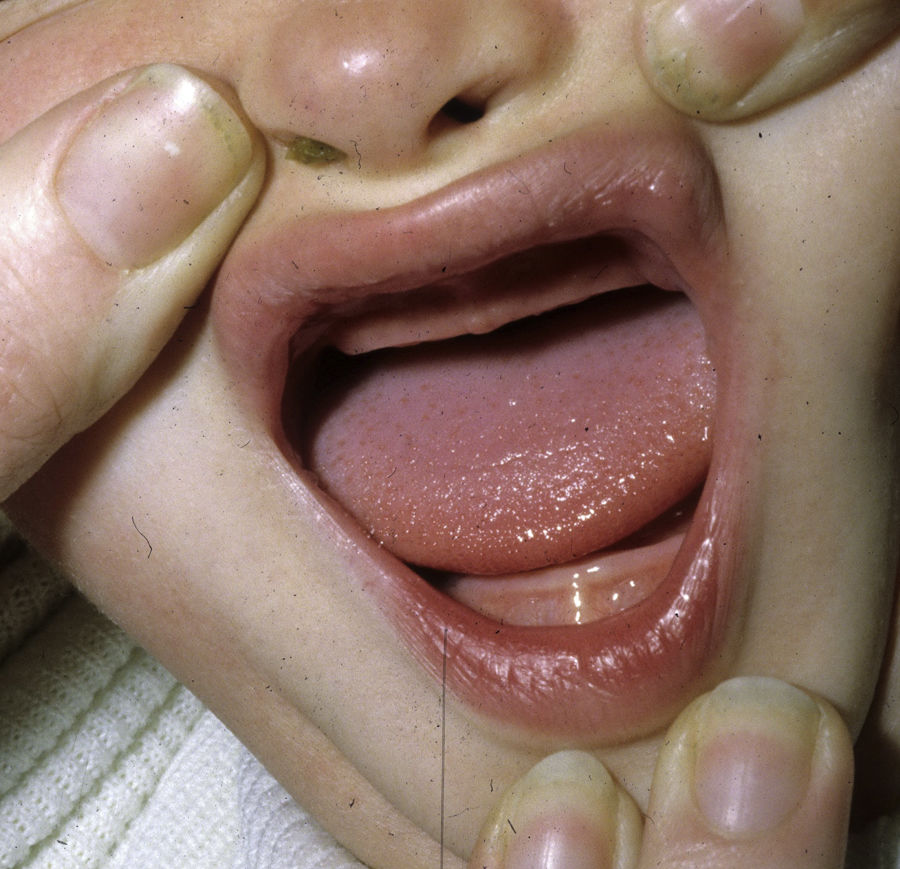
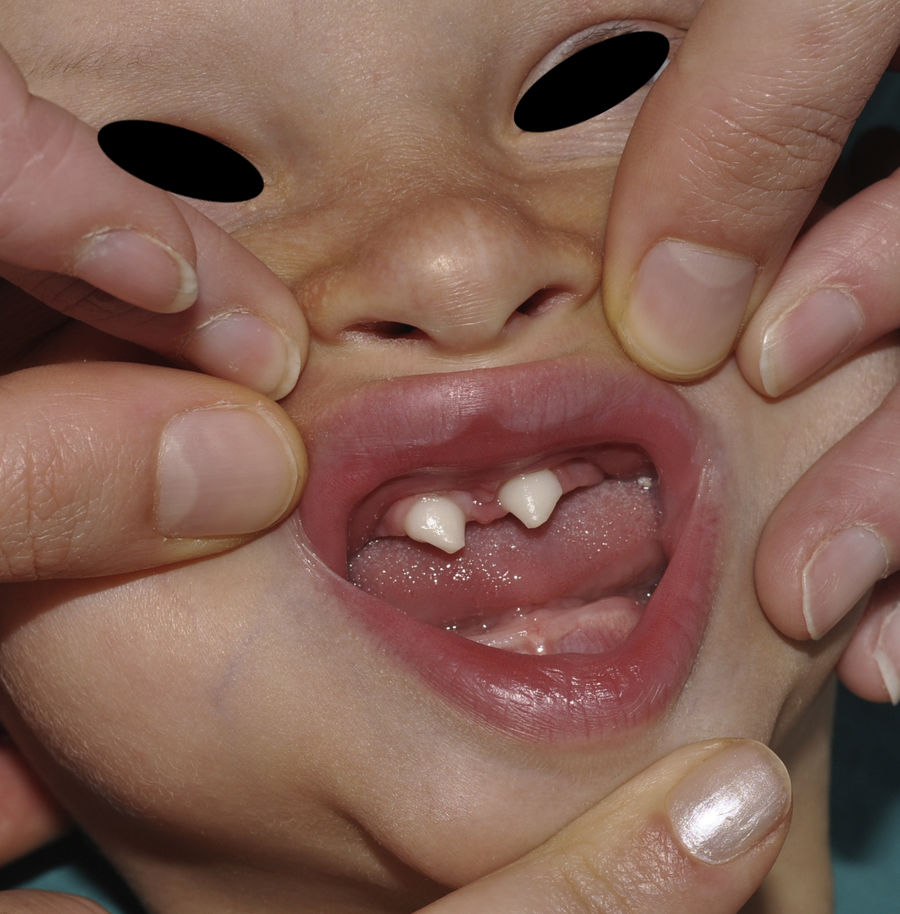
Los rasgos clínicos distintivos incluyen alteraciones en el pelo, los dientes y la sudoración. En el neonato las anomalías características son poco evidentes, por lo que el diagnóstico durante este periodo es difícil. Hasta el 70% de los niños varones con DEH-X presenta descamación de la piel en el periodo neonatal, hallazgo relativamente común también en las mujeres portadoras. Se han descrito casos de presentación como bebé colodión34. La alopecia puede ser la primera característica llamativa, pero raramente es total35 (fig. 3). La mayoría de los pacientes presentan cabello escaso, fino y rubio durante la infancia, que tiende a oscurecerse y hacerse más grueso con la edad. Las cejas y la barba son también escasas, pero las pestañas pueden ser normales, así como el vello axilar y pubiano. El resto del vello corporal también es escaso o incluso está ausente36. En algunos pacientes se observan alteraciones en el tallo del pelo, pero no son específicas de la DEH-X37. Las anomalías dentales pueden manifestarse durante la lactancia como hipoplasia de las crestas alveolares (fig. 4). Existe una notable variación inter e intrafamiliar en el número de dientes ausentes o malformados, así como importantes diferencias entre ambos sexos38. Las alteraciones morfológicas son más evidentes en los dientes anteriores, siendo lo más habitual una corona anómala, con forma de clavija o morfología cónica35 (fig. 5). Radiográficamente es común observar taurodontismo, anomalía que afecta principalmente a los molares y que se caracteriza por un acortamiento de la raíz sin alteración de la altura total del diente, el cual adquiere una forma prismática38. La capacidad de sudar está reducida o ausente, por lo que los pacientes son propensos a desarrollar hipertermia con los esfuerzos físicos o el calor ambiental36. Más del 90% de los niños presentan picos febriles recurrentes, sin otra causa explicable, durante el primer año de vida. Las convulsiones febriles aparecen en aproximadamente el 6% de los niños con DEH-X39. Los trastornos de la sudoración contribuyen sustancialmente a la morbilidad y mortalidad asociada a la DEH-X; así, el retraso mental cifrado en algunas series entre el 30-50% de los casos, pudiera ser debido al daño producido por la fiebre prolongada y las convulsiones febriles35. Aunque parece existir correlación geno-fenotípica en cuanto al grado de disfunción glandular, no es posible predecir el riesgo de hipertermia ni valorar la morbimortalidad de los pacientes40.



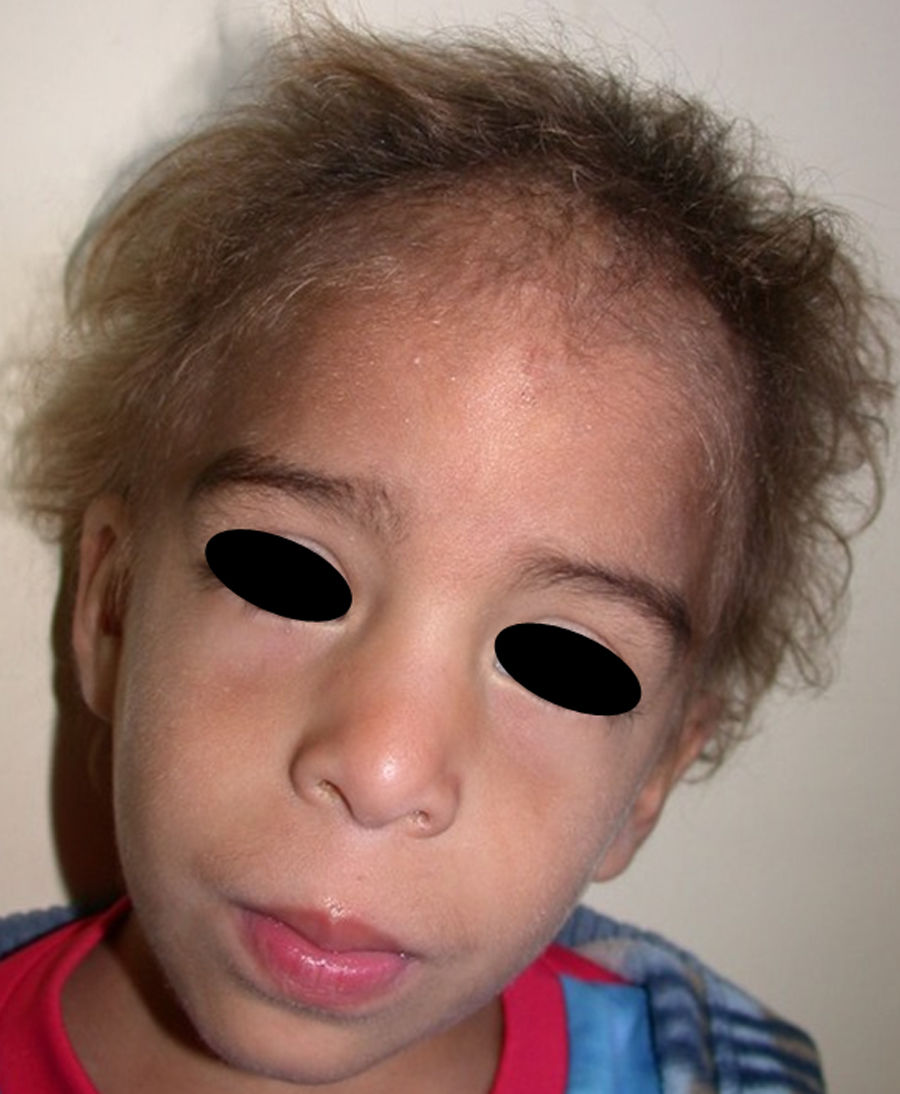
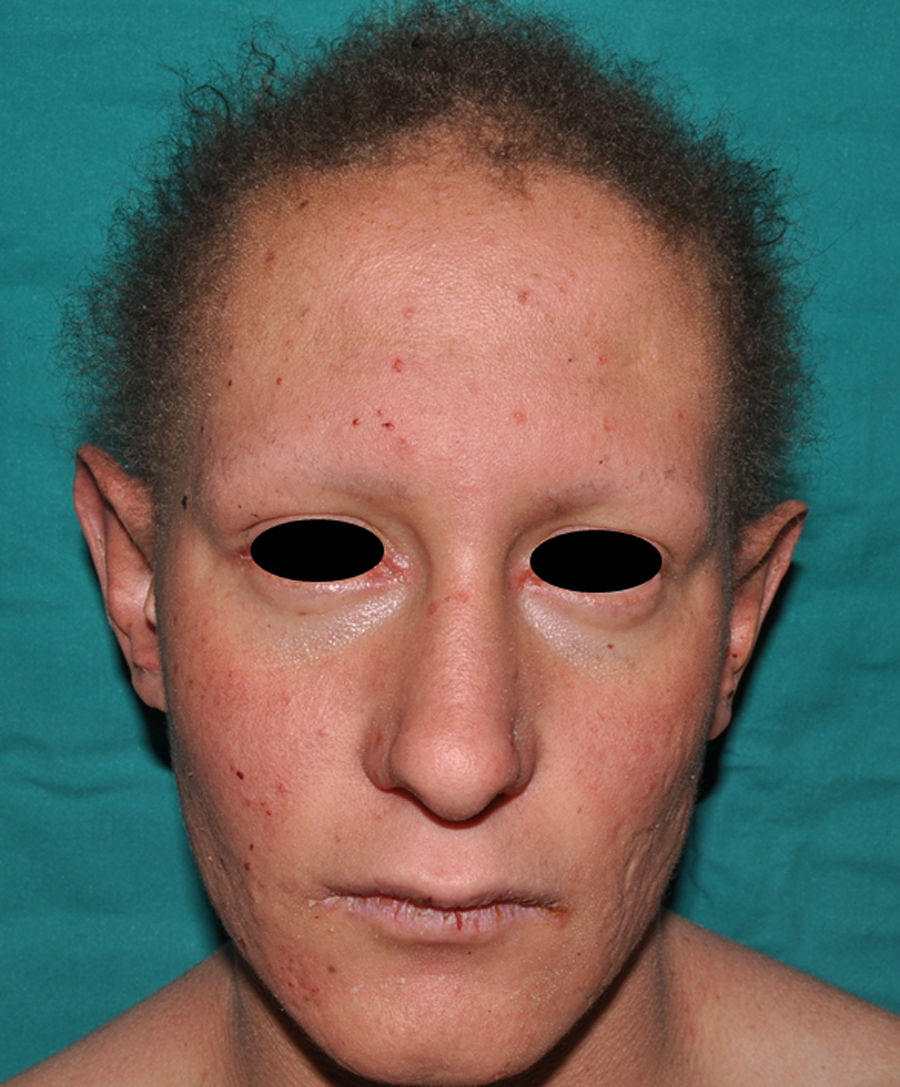
En las formas completas de la enfermedad la facies es peculiar, con prominencia frontal y de la barbilla, puente nasal hundido, labios gruesos y evertidos, orejas grandes y el hueso maxilar ancho y alto35. La mayoría de las alteraciones de la morfología craneofacial podrían explicarse por la ausencia de las piezas dentales, aunque también se postula que podrían ser debidas a la alteración de la morfogénesis embrionaria41. Además de los rasgos típicos, a partir de la niñez los pacientes suelen presentar piel seca, fina y brillante, hiperpigmentación periocular y finas arrugas perioculares que les confieren un aspecto de envejecimiento prematuro (fig. 6), así como pequeñas lesiones papulosas que remedan las hiperplasias sebáceas35. Hasta 2 tercios de los pacientes presentan eccema de tipo atópico, que puede ser de difícil manejo. Es raro encontrar queratodermia palmoplantar que, por el contrario, es un componente clásico de la displasia ectodérmica hidrótica (DEHi) o síndrome de Clouston42. Las uñas pueden ser normales o presentar fragilidad, pero no suelen ser excesivamente distróficas35.

La anomalía de las glándulas mucosas provoca unas secreciones nasales muy espesas que promueven el desarrollo de infecciones del tracto respiratorio35,36. La reducción de la secreción de las glándulas salivales podría, según algunos autores43, aumentar el riesgo de caries e infecciones orales fúngicas, así como dificultar la ingesta de alimentos y el habla. La reducción o ausencia de las glándulas de Meibomio, y la disfunción de las glándulas de Moll y Zeiss pueden provocar anomalías palpebrales a partir de la segunda década de la vida44; en los oídos el espesor del cerumen puede conducir a la obstrucción del conducto auditivo externo y condicionar una hipoacusia43. Las glándulas mamarias pueden ser hipoplásicas o incluso presentar agenesia total45, pero el desarrollo sexual es habitualmente normal35.
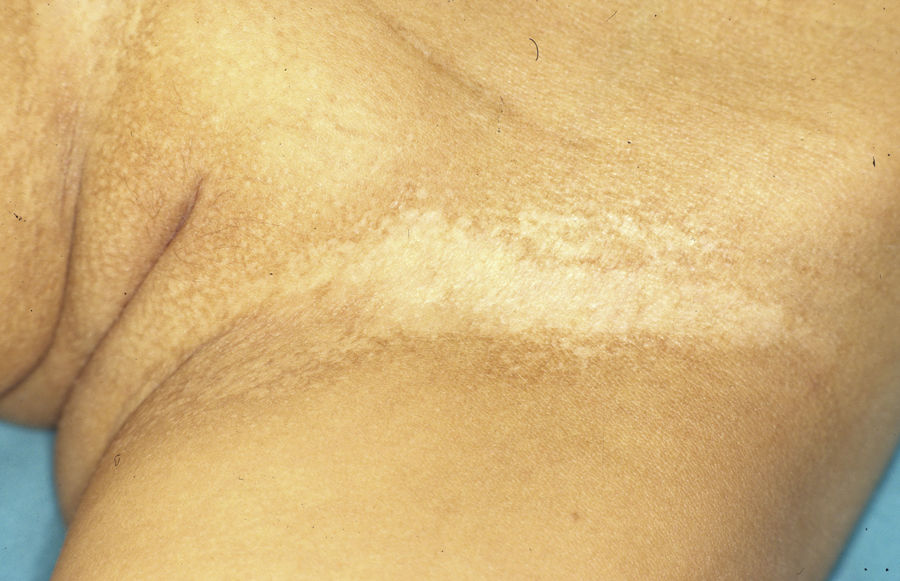
Las mujeres portadoras de un gen ED1 anómalo pueden ser asintomáticas o mostrar manifestaciones clínicas leves o moderadas46. Se ha observado que hasta el 70% de ellas muestra algún rasgo sugestivo de la enfermedad47. Las manifestaciones más frecuentes son las anomalías dentales, una ligera hipohidrosis y diferentes grados de hipotricosis46,48. Es posible encontrar una distribución parcheada del vello corporal, con áreas alopécicas, xeróticas y algo deprimidas de distribución blaschkoide que alternan con áreas normales de piel. Estas áreas afectadas se hacen más evidentes con el bronceado y durante la infancia46. En algunas portadoras se ha observado la desviación radial de la falange distal del dedo índice49.
Displasia ectodérmica anhidrótica de herencia autosómicaLas formas autosómicas de DEH, que son mucho menos frecuentes que las ligadas a X, pueden ser dominantes o recesivas, y se producen en la gran mayoría de los casos por mutaciones en los genes EDAR o EDARADD19. Desde el punto de vista clínico, los pacientes afectados por una DEH autosómica son clínicamente indistinguibles de los varones afectados por una DEH-X50,51. En general, los pacientes con la forma recesiva presentan anomalías más severas, mientras que el espectro clínico de las formas dominantes es variable52,53, y a veces es muy leve, prácticamente igual al que se observa en las portadoras de DEH-X26,53. Los portadores heterocigóticos en los patrones recesivos son clínicamente indistinguibles de los individuos genotípicamente normales54. La heterogeneidad genética de la DEH, y la similitud clínica entre pacientes con diferentes modos de herencia se explica por la implicación de la ectodisplasina, EDAR y EDARADD en la misma vía, ya que la activación de NFkB a través de la ectodisplasina es necesaria para la iniciación, formación y diferenciación de los derivados ectodérmicos54.
Diagnóstico de las displasias ectodérmicas anhidróticasEl diagnóstico precoz de las DEH es importante para prevenir las complicaciones en la etapa neonatal, derivadas fundamentalmente del control ineficaz de la temperatura corporal. El diagnóstico clínico es suficiente en las formas completas de la enfermedad, pero puede ser difícil en las formas incompletas y en los portadores de la enfermedad34,35. En estos casos es necesario realizar exploraciones complementarias para demostrar la disminución de la sudoración o el número de glándulas ecrinas.
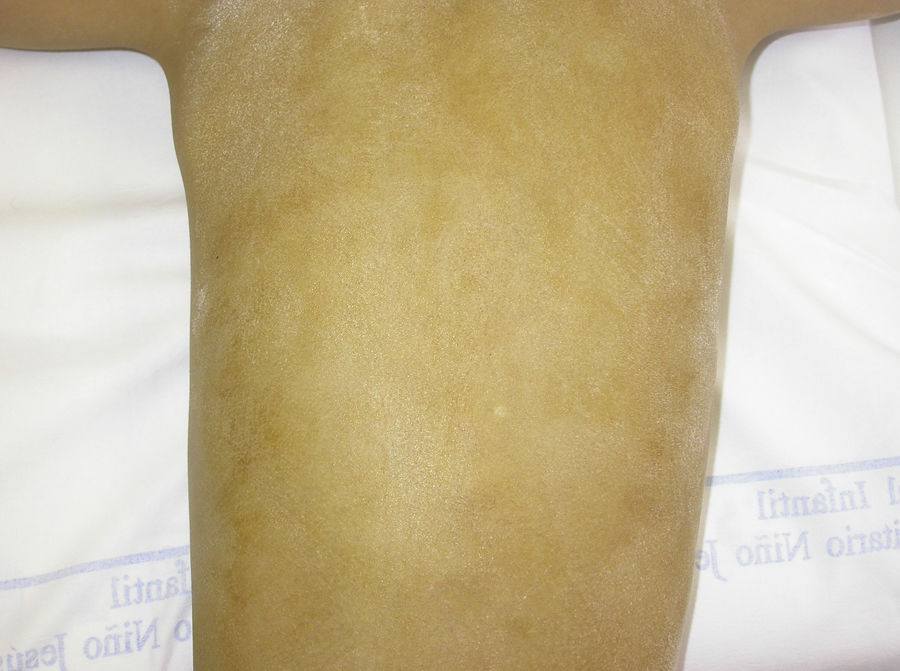
El test de Minor o de iodo-almidón es muy útil para confirmar la ausencia de sudoración, que será generalizada en los individuos afectados y parcheada en las mujeres portadoras, ya que estas poseen glándulas ecrinas normofuncionantes alternando con glándulas hipofuncionantes en áreas de distribución blaschkoide (fig. 7). El examen de grandes áreas del cuerpo, como la espalda, permite ver con mayor facilidad esta distribución en mosaico. Además, es una exploración útil para diferenciar a las portadoras de DEH-X de las mujeres afectadas por una DEH autosómica, en las que se observa una ausencia casi completa de la función de las glándulas sudoríparas46. Otros métodos de valoración de la sudoración incluyen la iontoforesis tras aplicar pilocarpina en el antebrazo, el recuento de los poros sudoríparos, el análisis de la conductancia de la piel o la termografía, métodos que suelen ser útiles como herramienta de screening, pero que tienen menor sensibilidad en los pacientes con función glandular residual40. La biopsia de piel normalmente no es imprescindible para la confirmación, pero la ausencia de glándulas ecrinas ofrece un valor predictivo positivo y una especificidad diagnóstica del 100%37. El análisis molecular es el único modo de determinar el gen implicado, de detectar portadores y de confirmar el patrón de herencia, algo imprescindible para ofrecer consejo genético.
Pronóstico y tratamiento
Hace años se creía que la tasa de mortalidad de los niños con DEH-X alcanzaba el 30% durante la primera infancia43, pero hoy se sabe que el riesgo es menor, en torno a un 13%. Las complicaciones aparecen en los primeros años de la vida debido a la hipertermia y a las infecciones respiratorias, pero una vez pasada la niñez la esperanza de vida es normal39. Educar al paciente y a su familia es esencial, ya que deberán evitar situaciones de hipertermia. No es necesario eliminar por completo la actividad física, pero deben conocer los síntomas derivados del exceso de temperatura corporal, como la cefalea, las náuseas y vómitos, la sensación de mareo, el cansancio excesivo o los calambres musculares, así como las técnicas para disminuir la hipertermia (inmersión en agua, aire acondicionado, bebidas frías, empleo de artilugios refrigerantes, etc.). Los deportes acuáticos son ideales para estos pacientes.
No existe tratamiento para los trastornos del pelo ni para la hiperpigmentación periocular, y los brotes de dermatitis atópica pueden ser difíciles de tratar. Algunos autores han sugerido un incremento en el riesgo de presenter melanoma, por lo que se recomienda hacer una exploración física completa anual en estos pacientes55. El manejo de los niños con DEH incluye, además, el cuidado dental precoz para prevenir la hipoplasia maxilar y la atrofia de las encías, que en los casos más severos pueden dificultar la masticación y el desarrollo del lenguaje, y a menudo son causa de un notable problema estético. Otros especialistas involucrados en el cuidado de estos pacientes son el otorrinolaringólogo cuando existen problemas con la secreción nasal y el cerumen ótico, el oftalmólogo si existe sequedad ocular o problemas palpebrales, el neumólogo en caso de infecciones del tracto respiratorio asociadas y, en algunos casos, el psicólogo36. La terapia génica con EDA recombinante todavía está en fase experimental, pero parece ofrecer un futuro esperanzador para estos pacientes56–58. Finalmente, y al igual que ocurre en otras muchas enfermedades genéticas, los pacientes diagnosticados precisarán tanto información continua sobre su enfermedad como apoyo socioeconómico, por lo que nosotros les recomendamos que contacten con la asociación española de pacientes con DEH (AADE): http://www.displasiaectodermica.org/.
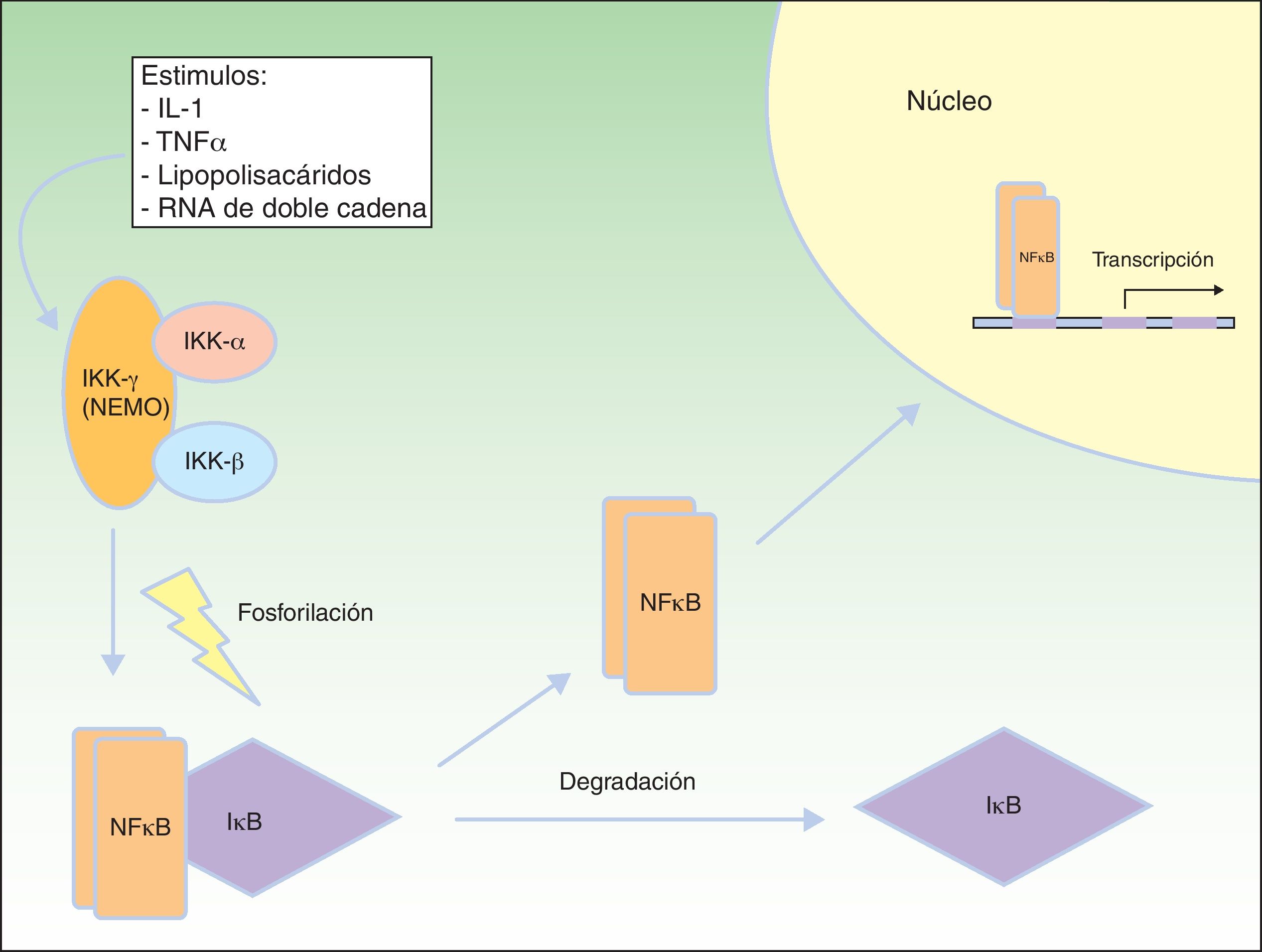
Vía de señalización del factor nuclear kBEl factor nuclear kB (NFkB) es un factor de transcripción que regula la expresión de múltiples genes implicados en las respuestas inmune e inflamatoria, así como en la reacción frente al estrés, en la adhesión celular y en la protección frente a la apoptosis59–61. En la mayoría de las células el NFkB se encuentra inactivado mediante secuestro citoplásmico por la proteína inhibidora de NFkB (IkB). Varios estímulos, como la interleucina 1 (IL-1), el TNFα, los lipopolisacáridos (endotoxinas bacterianas) y el ARN de doble cadena (infecciones virales) producen la activación de receptores de membrana celular pertenecientes a la familia del TNF, que incluyen EDAR y el receptor activador de NFkB (RANK)62. La activación de estos receptores conduce a la degradación de IkB mediante el complejo IKK que fosforila a IkB y permite la translocación de NFkB dentro del núcleo, donde induce la transcripción génica, desencadenando respuestas inflamatorias e inmunes críticas en el desarrollo de las células T y B, la función de los osteoclastos y el crecimiento de las células epidérmicas63. El complejo IKK consta al menos de 3 subunidades: IKK1/IKKα, IKK2/IKKβ y NEMO/IKKγ (modulador esencial del factor nuclear kB). IKK1 e IKK2 actúan como subunidades catalíticas, mientras que NEMO es una subunidad estructural y reguladora, esencial para que el complejo funcione como una unidad. La ausencia de NEMO resulta en la no actividad de NF-kB en respuesta a los estímulos64,65, ya que NEMO es la principal molécula que proporciona esta señalización desde el núcleo al citoplasma66 (fig. 8).
 es el componente regulador del complejo IKK. Ante diferentes estímulos se activa y fosforila a IκB, produciendo su degradación y la liberación de NFκB, que viaja al núcleo celular donde activa genes de transcripción. Modificada de Nelson DL73.")
Vía NEMO y NFkB. NEMO (IKKγ) es el componente regulador del complejo IKK. Ante diferentes estímulos se activa y fosforila a IκB, produciendo su degradación y la liberación de NFκB, que viaja al núcleo celular donde activa genes de transcripción. Modificada de Nelson DL73.
Tanto la activación como la inhibición de NFκB se han asociado con el desarrollo de enfermedades inflamatorias cutáneas67, y se ha demostrado que las mutaciones en 2 genes, NEMO e IkBα, originan un grupo heterogéneo de trastornos genéticos entre los que se incluye la incontinencia pigmenti (IP), la DEH con inmunodeficiencia ligada a X (DEH-ID-X), la DEH con inmunodeficiencia, osteopetrosis y linfedema (DEH-ID-OL), y la DEH con inmunodeficiencia de herencia autosómica dominante (DEH-ID-AD) entre otros trastornos61,68,69.
Incontinencia pigmentiEs una enfermedad infrecuente, de prevalencia no bien conocida, causada por una alteración molecular en el gen NEMO, el cual se localiza en el cromosoma X (locus Xq28)70,71. En torno al 97% de los individuos afectados son mujeres, ya que es letal intraútero para la mayoría de los varones afectos. La mayoría de las mujeres sobreviven debido a la eliminación selectiva de las células que expresan el cromosoma X que presenta la mutación72, siendo en ellas la expresividad clínica variable70,73. No obstante, los varones también pueden padecer la enfermedad en caso de mosaicismo somático o trisomía XXY65,74,75.
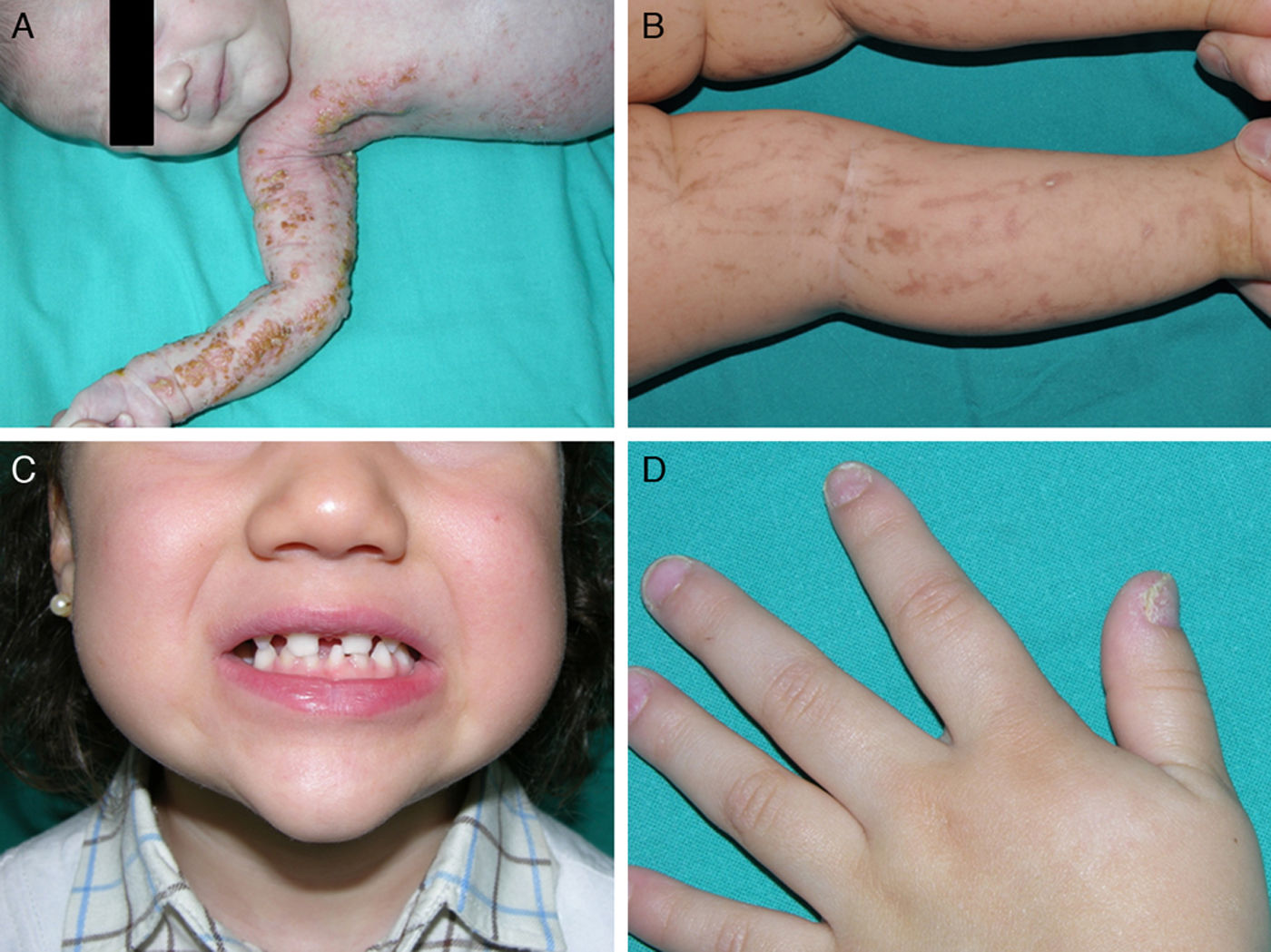
Las manifestaciones cutáneas son las más llamativas, pero no las más graves; están presentes en casi el 100% de las pacientes afectadas y aparecen en la etapa neonatal. Las lesiones se distribuyen siguiendo las líneas de Blaschko y clásicamente se dividen en 4 estadios: vesicular, verrucoso, hiperpigmentado y atrófico65 (fig. 9). Ocasionalmente, los primeros estadios pasan desapercibidos, y las pacientes solo muestran discretas lesiones hiperpigmentadas que pasan desapercibidas hasta que dan a luz una hija afectada. Algunas mujeres, incluso, no presentan manifestaciones a pesar de portar la mutación73. Los problemas clínicos de mayor relevancia son los trastornos visuales y las alteraciones neurológicas, pero afortunadamente son menos frecuentes que las manifestaciones dermatológicas, apareciendo en el 40 y en el 30% de los casos respectivamente76. Además, las pacientes pueden presentar otros problemas como alopecia, alteraciones dentales (dientes cónicos o en clavija, hipodontia o anodontia) y distrofia ungueal73. Puesto que la revisión profunda de este trastorno se encuentra fuera de nuestros objetivos, remitimos al lector interesado a consultar otras fuentes bibliográficas.

Incontinencia pigmentaria: distintos aspectos clínicos. A. Estadio vesiculoso en un neonato. B. Lesiones lineales hiperpigmentadas en las extremidades inferiores en un estadio más tardío. C. Agenesia dental y presencia de dientes cónicos. D. Distrofia ungueal, con traquioniquia y piqueteado de la placa ungueal.
Es una rara enfermedad de herencia recesiva ligada a X que afecta mayoritariamente a varones77, aunque se han descrito algunos casos en el sexo femenino78,79. La incidencia estimada es de 1 por cada 250.000 recién nacidos80. La mayoría de los pacientes presentan pequeñas deleciones o mutaciones nonsense en el dominio zinc finger (zona ligada a cinc que permite la interacción con otras moléculas) de NEMO, que no provocan una pérdida completa de la activación de NFkB, como ocurre en la IP, sino una función alterada o disminuida de la vía NF-kB77.
Tanto los varones como las mujeres con DEH-ID-X tienen madres con lesiones cutáneas que recuerdan a la incontinencia pigmenti, así como manifestaciones variables de DEH, entre las que se incluyen los dientes cónicos81,82. Algunos pacientes presentan prominentes venas superficiales83. También se ha descrito un fenotipo cutáneo con afectación inicial de las áreas intertriginosas de aspecto seborreico que progresa a eritrodermia y que recuerda al de otros pacientes con inmunodeficiencias congénitas como la inmunodeficiencia combinada severa o el síndrome de Omenn84. En algún caso se ha observado lesiones de IP, demostrándose la complejidad y solapamiento entre las diferentes enfermedades derivadas de la alteración de NEMO81. Los pacientes con DEH-ID-X presentan una pobre respuesta inflamatoria secundaria a la alteración de la respuesta celular frente a citoquinas proinflamatorias (IL-1beta, IL-18 y TNF-alfa)60. La inmunodeficiencia más común es una disgammaglobulinemia con niveles normales o bajos de IgG, pero también pueden existir niveles elevados de IgA, IgM e IgE85. Además se han descrito defectos en la activación de las células natural killer (NK), una disminución en la producción de TNF e IL-12, una respuesta anómala a la estimulación por polisacáridos con incapacidad para formar anticuerpos específicos frente a Streptococcus pneumoniae y una producción retrasada o ausente de isohemaglutininas77,85,86. Por todo ello, además del característico fenotipo de la DEH, estos pacientes presentan infecciones bacterianas serias y recurrentes en el tracto respiratorio bajo, la piel y los tejidos blandos, los huesos, el tracto gastrointestinal y las meninges65,87. Los gérmenes patógenos son habitualmente bacterias piógenas como Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Klebsiella, Salmonella y Pseudomonas spp., así como micobacterias, citomegalovirus, virus herpes simple y Pneumocystis jiroveci80. Aunque las complicaciones infecciosas son consideradas generalmente las más amenazantes para la vida, los defectos de señalización de NF-kB también aumentan el riesgo de enfermedades inflamatorias, en particular de colitis inflamatoria80.
Las mujeres portadoras tienen manifestaciones diversas, desde una dentadura normal a ligera hipodontia o dientes cónicos. Anecdóticamente se ha descrito el caso de 2 mujeres que presentaron pigmentación moteada de la piel77.
Displasia ectodérmica hipohidrótica con inmunodeficiencia autosómica dominanteEn los casos de herencia dominante los pacientes presentan una mutación de IkBα que impide la fosforilación y degradación de la proteína IkBα, resultando en una activación alterada de NFkB88. Estos pacientes presentan las características clásicas de la DEH, junto con una inmunodeficiencia de células T que da lugar a infecciones recurrentes y defectos inmunológicos89. Aunque los pacientes con mutaciones en NEMO e IκBα comparten algunas manifestaciones clínicas, sus fenotipos inmunológicos son distintos90. Estos pacientes presentan una profunda deficiencia de células T caracterizada por la pérdida de células CD45RO+ y la alteración de la estimulación de los linfocitos vía receptor de células T in vitro, con incapacidad para responder a la estimulación por el TNFα, presentando una producción de anticuerpos anómala y una mayor susceptibilidad a las infecciones por bacterias grampositivas y gramnegativas, de manera similar a lo que ocurre en los pacientes con mutaciones de NEMO. Sin embargo, estos pacientes tienen una actividad normal de las células NK, por lo que no presentan infecciones por micobacterias89. Parece que mutaciones hipomórficas en el codón de parada (stop codon) de IkBα podrían producir una menor alteración de la activación de NF-kB, dando lugar a una inmunodeficiencia menos grave88.
Las mutaciones en el stop codon del gen NEMO resultan en la DEH-ID-OL77. Al igual que los niños con DEH-ID-X, estos presentan manifestaciones cutáneas de DEH, pueden presentar lesiones cutáneas similares a la IP, y tienen anomalías en la respuesta inflamatoria85,91. La inmunodeficiencia es particularmente severa, por lo que sufren infecciones inusuales y agresivas desde la primera infancia, frecuentemente mortales60,65. Las características clínicas distintivas son la osteopetrosis y el linfedema, que se creen debidas a una alteración en la señalización de RANK, un receptor de la familia del TNF presente en las células progenitoras de osteoclastos87,92, cuya diferenciación y funcionamiento dependen de NF-kB69. En estos pacientes la diferenciación osteoclástica está abolida o severamente disminuida, lo cual determina la aparición de unos huesos densos pero frágiles92. Por otro lado, el gen NEMO codifica al receptor 3 del factor de crecimiento del endotelio vascular (VEGFR3), un activador de la vía NFkB93 cuya señalización se vería interferida, dando lugar a una disfunción de los vasos linfáticos y el linfedema característico69.
Las mujeres portadoras de una mutación hipomórfica en el gen NEMO pueden ser asintomáticas o tener varias manifestaciones de incontinencia pigmentaria69,94.
Diagnóstico de los trastornos derivados de NEMO/IkBαLas mutaciones del gen NEMO deben tenerse en cuenta en niños con una extensa dermatitis de tipo seborreico o atópico de curso recalcitrante, particularmente cuando concurren rasgos faciales de DEH o hay historia materna de IP84. Se recomienda biopsiar las erupciones cutáneas presentes en los pacientes con DEH-ID ya que pueden desarrollar lesiones similares a la IP después de la infancia81.
TratamientoEl manejo de las manifestaciones de DEH es similar al comentado en el apartado anterior. El tratamiento de la inmunodeficiencia puede incluir terapias inmunes y el manejo agresivo de las infecciones, incluyendo profilaxis frente a bacterias grampositivas y gramnegativas, micobacterias, citomegalovirus, virus herpes simple y Pneumocystis jiroveci80. Con todo, la morbilidad y la mortalidad son significativas77. El trasplante de células hematopoyéticas ofrece la posibilidad de la reconstitución inmune, pero conlleva los riesgos inherentes a la inmunosupresión69. Aunque hay pocos datos publicados al respecto, parece que el trasplante alogénico de progenitores hematopoyéticos podría corregir la inmunodeficiencia asociada a las enfermedades por mutación de NEMO o IkBα; sin embargo, las mutaciones causantes de la enfermedad no son exclusivamente hematopoyéticas, por lo que las manifestaciones constitucionales no inmunes no se corrigen con el transplante, e incluso parece que la colitis inflamatoria podría empeorar al corregir la inmunodeficiencia95.
Alteración de reguladores de la transcripción y/o expresión de determinados genesTrastornos derivados de mutaciones en p63El gen p63, también conocido como TP63 (tumor protein 63), se localiza en el cromosoma 3 (locus 3q27) y codifica el factor de transcripción p63, involucrado en el desarrollo ectodérmico. Las regiones de mayor importancia biológica son el dominio DNA binding domain (zona que permite la transactivación mediante la unión de la proteína p63 con el ADN), el dominio Sterile Alpha Motif (SAM), que se cree que participa en la interacción entre proteínas, y el dominio de inhibición de la transactivación (TID), localizado al lado del dominio SAM y que podría tener un papel representativo en equilibrar los efectos de diferentes isoformas de TP6396. La proteína p63 se expresa muy tempranamente durante la embriogénesis y desempeña un papel esencial en la inducción de la diferenciación y proliferación de la epidermis, así como en otros procesos, incluyendo el desarrollo facial y de los miembros96–98. Es posible que la ausencia de expresión de la misma durante el desarrollo temprano de las estructuras ectodérmicas bloquee una cadena de interacciones entre epitelio y mesénquima, interfiriendo la morfogénesis normal99. Además, p63 regula la expresión de P-cadherina, un regulador crítico del desarrollo del pelo100.
Mutaciones heterocigóticas en el gen p63 son responsables de al menos 6 síndromes diferentes que combinan DE, hendidura orofacial y malformaciones de los miembros101. El síndrome ectrodactilia-displasia ectodérmica-labio/paladar hendido (EEC) es el prototipo de estos síndromes, entre los que también se encuentran el anquilobléfaron-displasia ectodérmica-labio/paladar hendido (AEC), limb-mammary syndrome (LMS), acro-dermato-ungual-lacrimal-tooth syndrome (ADULT), el síndrome Rapp-Hodgkin (RHS) y la split-hand/foot malformation (SHFM4). Todos ellos presentan al menos una de las características claves del síndrome EEC101. Existe una fuerte correlación geno-fenotípica que depende de la localización de la mutación en p6396. Los residuos aminoácidos más frecuentemente mutados son R204, R227, R279, R280 y R304. El fenotipo relacionado con la mutación en R204 es muy similar al fenotipo completo del síndrome EEC, pero los pacientes presentan menor frecuencia de hipohidrosis y hendidura orofacial; las mutaciones del aminoácido R227 se asocian raramente con hendidura orofacial y sindactilia, pero tienen mayor incidencia de problemas renales e hipohidrosis, así como ausencia de alteraciones auditivas; la mutación en R279 es la única que origina anquilobléfaron, y suele asociarse a ectrodactilia (deformidad de las extremidades en la que hay ausencia de partes o dedos completos, adoptando las manos o los pies una forma que recuerda a las pinzas de una langosta); los pacientes con mutación en R280 presentan frecuentes signos cutáneos y sindactilia, pero no hipohidrosis ni problemas auditivos o renales; por último, la mutación en R304 se relaciona con un alto porcentaje de hendidura orofacial, sindactilia y alteraciones auditivas101.
Síndrome ectrodactilia-displasia ectodérmica-labio/paladar hendidoEl síndrome EEC es relativamente común. Aunque pueden aparecer casos esporádicos, la mayoría presentan una herencia autosómica dominante102. El 98% de los pacientes con el fenotipo clásico presenta mutaciones en el gen p63, generalmente mutaciones puntuales localizadas en el dominio de unión al ADN, y solo ocasionalmente en los dominios SAM y TID96. Se han identificado al menos 30 mutaciones diferentes, de las cuales 5 (las mutaciones en los aminoácidos R204, R227, R279, R280 y R304 de p63) son responsables del 86% de los casos101.
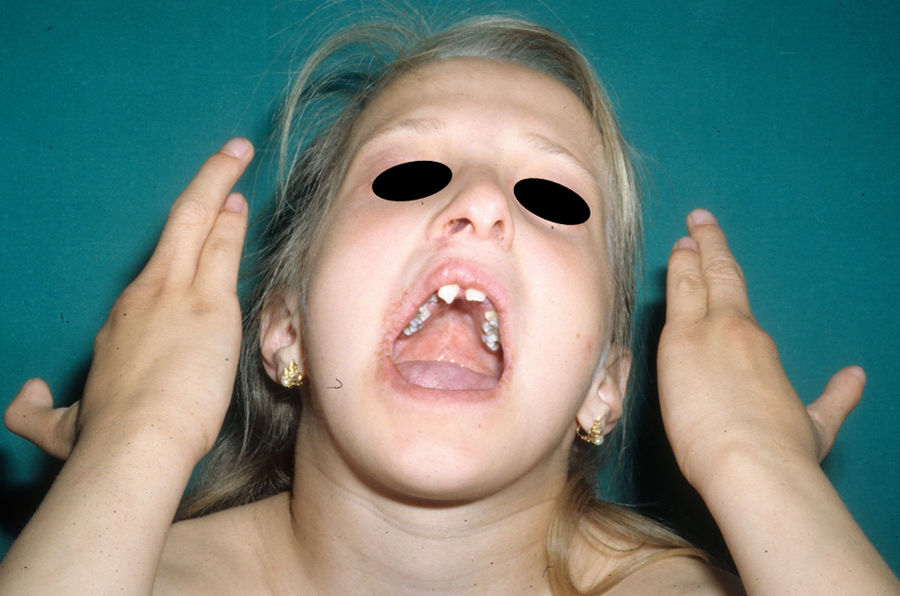
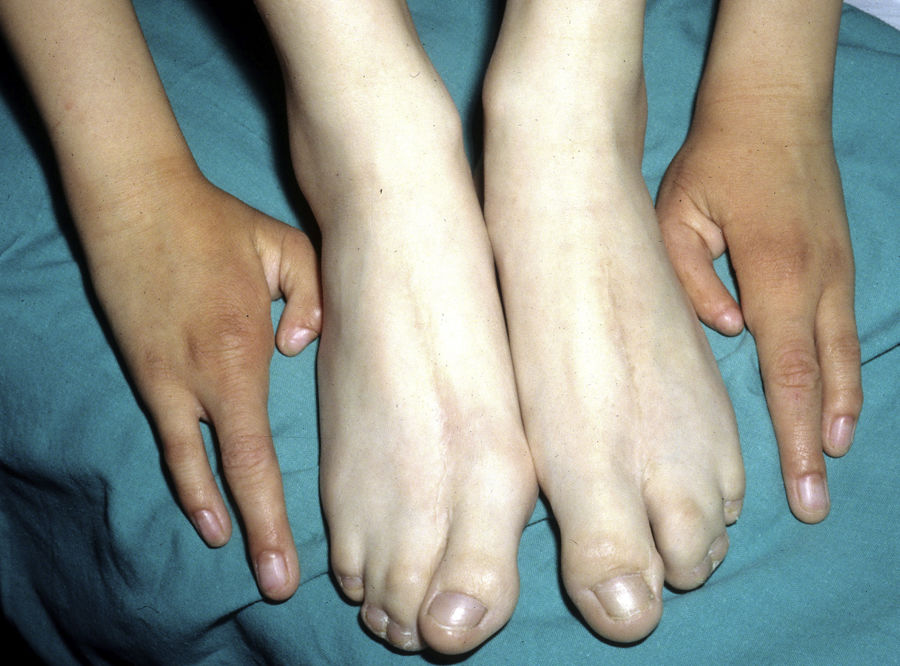
Las alteraciones más frecuentes son las malformaciones en los miembros, la DE y la hendidura orofacial (fig. 10), seguidas de las anomalías del conducto lacrimal, malformaciones genitales y la sordera, aunque existe una gran variabilidad clínica intra e interfamiliar103. Entre las alteraciones de los miembros las más representativas son la ectrodactilia y la sindactilia (fig. 11). Como signos de displasia ectodérmica pueden presentar pelo escaso, hipopigmentado, ausencia de cejas y pestañas y alopecia. La piel suele ser fina, seca y de aspecto atópico, y las uñas son distróficas y pueden mostrar piqueteado. Se han descrito lesiones periorales y queilitis angular en las comisuras orales de estos pacientes, que pudieran ser debidas a los cambios anatómicos provocados por la cirugía reconstructiva realizada para el labio/paladar hendido104,105. También se han descrito alteraciones dentales como hipodontia o anodontia, así como propensión a la caries debido a un defecto en el esmalte y una alteración en la función de las glándulas salivares. La hendidura orofacial es frecuente y se puede acompañar de hipoplasia maxilar y malar. La estenosis del conducto lacrimal contribuye a la queratitis, que a veces se asocia a fotofobia101. También puede haber anomalías uro y anogenitales (micropene, hipospadias, septo vaginal e hipoplasia de los genitales femeninos), insuficiencia hipotálamo-hipofisaria, hipoplasia tímica o retraso mental106,107. Se han descrito casos aislados de asociación con nevus blanco esponjoso108, así como con micrognatia, fisura del paladar blando y glosoptosis (secuencia de Pierre Robin), pero se desconoce la relevancia de dicha asociación109.


El diagnóstico prenatal mediante ecografía desempeña un importante papel en el diagnóstico del síndrome EEC, no solo porque la detección de ectrodactilia y labio/paladar hendido pueden alertar al clínico, sino también para la detección de anomalías genitourinarias graves asociadas107.
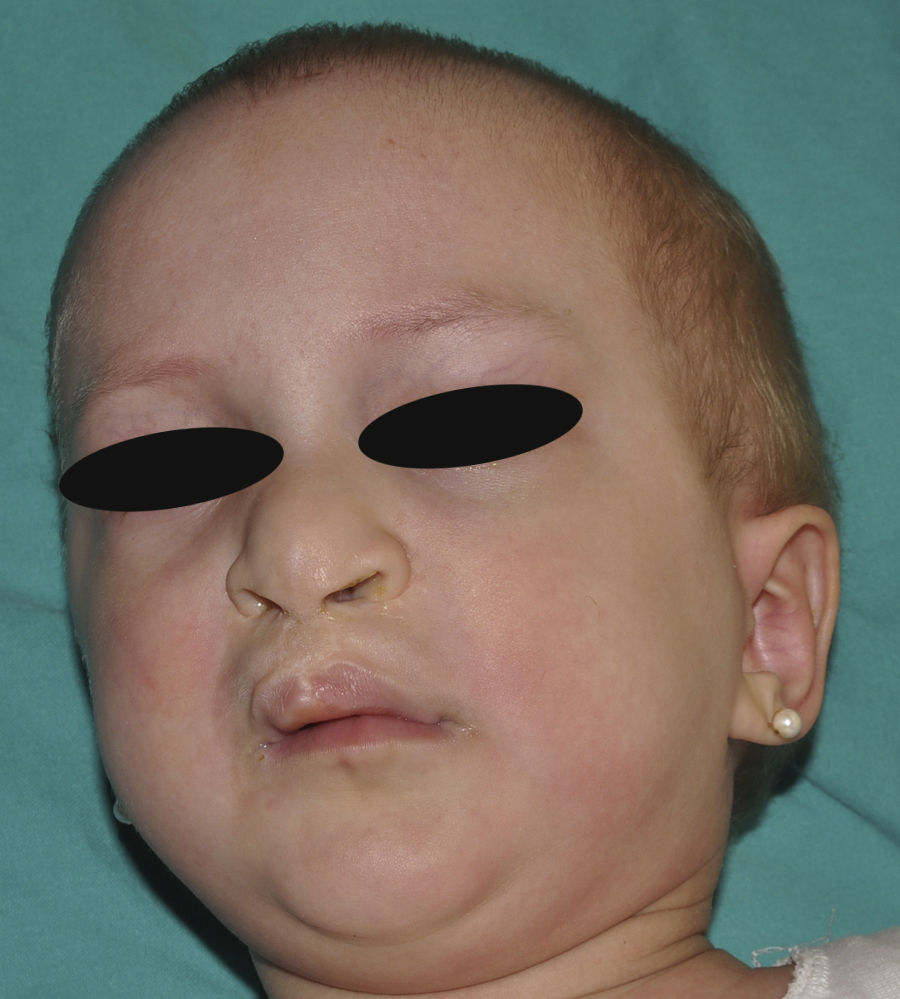
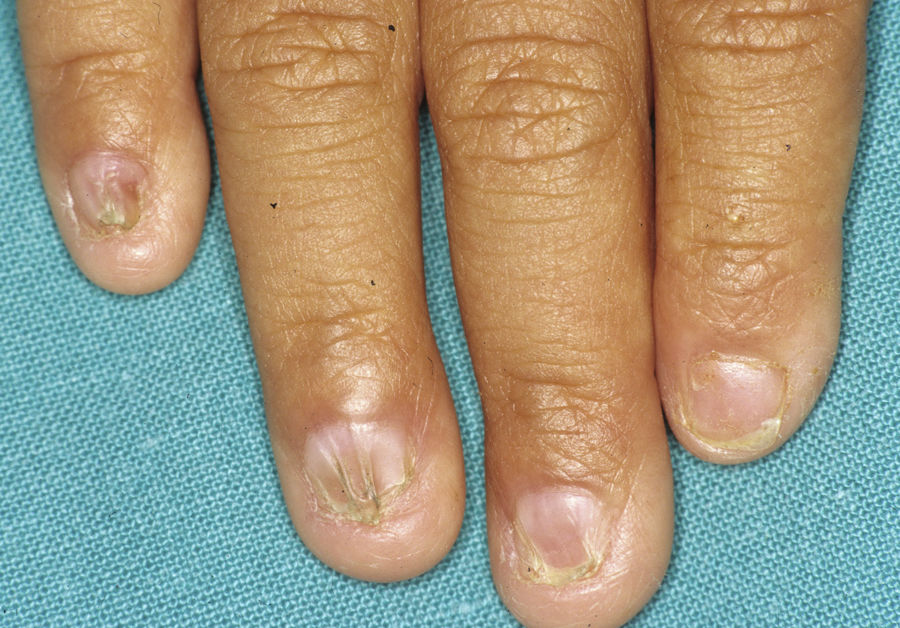
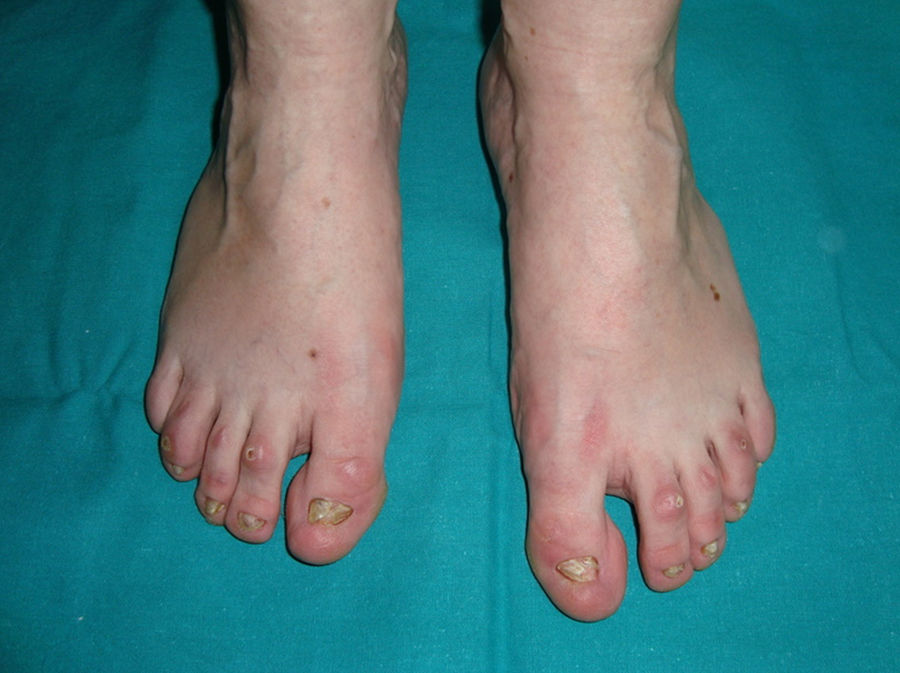
Síndrome anquilobléfaron-displasia ectodérmica-labio/paladar hendidoEl trastorno AEC es un cuadro autosómico dominante que se produce como resultado de mutaciones sin sentido en el dominio SAM de la proteína p63, implicado en la interacción con otras proteínas involucradas en la regulación de la transcripción y del desarrollo de los anejos cutáneos. La tríada clínica característica consiste en anquilobléfaron, defectos ectodérmicos y labio/paladar hendido (fig. 12)110. El anquilobléfaron consiste en la presencia de bandas fibrosas entre los párpados que impiden su apertura y movimiento normal. Las anomalías ectodérmicas son similares a las de otras DE96, y junto a las mismas pueden observarse erosiones cutáneas en el cuero cabelludo, la cabeza, el cuello, los pliegues, las palmas y las plantas que cicatrizan con dificultad y tienden a la sobreinfección. En la parte superior del tronco curan dejando una cicatrización residual de patrón cribiforme, reticulado, estrellado o punteado. Aunque se desconocen las razones exactas de las erosiones y de la pobre curación de las heridas, el papel de la proteína p63 en la formación de las células basales y la diferenciación de la epidermis podría contribuir111. En el momento del nacimiento los pacientes pueden presentar lesiones erosivas, eritrodermia congénita, descamación ictiosiforme e incluso membrana colodión, manifestaciones que inicialmente hacen pensar en una epidermólisis ampollosa o un trastorno de la queratinización111. Las alteraciones de la pigmentación también son frecuentes, tanto hipo como hiperpigmentación (fig. 13). En los grandes pliegues se observa una hiperpigmentación reticulada que se acentúa con la edad, mientras que en la cara se observa una característica hipopigmentación periocular con aspecto de máscara que mejora con el paso de los años. Prácticamente todos los pacientes presentan alteraciones de la sudoración111. El grado de alopecia es variable, y no parece correlacionarse con la edad o con el grado de erosiones previas del cuero cabelludo. El cabello puede ser grueso, hirsuto, frágil, o con aspecto deslustrado, y presenta pigmentación variable e incluso bitonalidad (algunos cabellos pigmentados y otros pálidos). Son frecuentes las alteraciones del tallo del pelo, incluyendo pili annulati, pili torti, pili canaliculi o identaciones irregulares, entre otros112. La afectación ungueal también es variable (fig. 14) y abarca desde la anoniquia completa a la leve descamación de la placa ungueal111. Otras anomalías cutáneas ocasionales incluyen el borramiento de los dermatoglifos, la hiperqueratosis palmoplantar, la queratodermia punctata, la hiperlinealidad y la hiperqueratosis en codos y rodillas111.
.")


La dismorfia facial incluye, además del paladar y el labio hendidos, orejas pequeñas o malformadas, hipoplasia maxilar, ausencia de dientes permanentes y disminución de la longitud de la hendidura palpebral. También es habitual encontrar retraso estatoponderal, sindactilia, atresia de los conductos naso-lacrimales, otitis media recurrente, pérdida auditiva e hipospadias96. Algunos individuos presentan neutropenia de etiología indeterminada y pueden tener infecciones cutáneas y óticas recurrentes que pueden progresar a bacteriemia y sepsis96.
Cualquier neonato con eritrodermia y presencia de labio/paladar hendido debe hacer sospechar un síndrome AEC, especialmente si existen otros signos cutáneos como las erosiones en el cuero cabelludo o en la piel, aunque el anquilobléfaron no siempre está presente. La biopsia de piel, poco específica, muestra leve hiperqueratosis, atrofia epidérmica, pigmentación basal y/o incontinencia pigmentaria, y un plexo vascular superficial prominente con un ligero infiltrado perivascular de predominio linfocítico112.
El primer objetivo del tratamiento es prevenir las erosiones cutáneas, por lo que se debe evitar la limpieza vigorosa de la piel. La infección secundaria de las heridas puede dificultar su curación, por lo que es recomendable extremar la higiene diaria y aplicar antisépticos en las erosiones. La administración de antibióticos durante largos periodos parece que únicamente consigue una mejoría mínima o temporal de las lesiones. Algunos autores han sugerido que el empleo de corticoides113,114 o las dosis bajas de doxiciclina115 pueden ser útiles para reducir la inflamación y mejorar la cicatrización, pero debe valorarse su empleo por la posibilidad de efectos secundarios. Futuras estrategias terapéuticas están bajo investigación, incluyendo terapia génica, así como el empleo de células madre epidérmicas para regenerar la piel afectada111.
Síndrome acro-dermato-ungueal-lacrimal-dentalLos síndromes EEC y ADULT son considerados trastornos alélicos con rasgos clínicos que se solapan como las anomalías ectodérmicas, las anomalías de los miembros y las alteraciones dentales. La principal diferencia es la hendidura orofacial, que está ausente en el síndrome ADULT116. Este síndrome se produce por mutaciones que afectan al dominio TID, implicado en la transactivación del gen p63117. Hasta el momento se han descrito 5 mutaciones de este gen, 2 de las cuales se han identificado también en pacientes con síndrome EEC118.
Síndrome de miembros y mamasEste síndrome, de herencia autosómica dominante, se produce por mutaciones en los dominios SAM y TID del gen p63119. Sus características clínicas se solapan con las de los síndromes EEC, AEC, ADULT y Rapp-Hodgkin, así como con el síndrome cúbito-mamario (causado por mutaciones en el gen TBX3-locus 12q24.1)120. Se caracteriza por ectrodactilia, hipoplasia de las glándulas mamarias y de los pezones, paladar hendido (sin labio hendido) y ausencia de anomalías en el pelo o la piel. Otras manifestaciones incluyen estenosis del conducto lacrimal, pérdida auditiva, anomalías urogenitales, displasia de la nariz, hipohidrosis, hipodontia y displasia gonadal99,120.
La diferenciación entre LMS y EEC se basa en 3 hallazgos120:a) la hipoplasia de las glándulas mamarias y pezones, presentes en todos los casos de LMS, y solo ocasionalmente en el EEC;b) las alteraciones del pelo y la piel, ausentes en los pacientes con LMS; y c) la presencia de labio hendido, que solo presentan los pacientes con síndrome EEC.
Síndrome de Rapp-HodgkinSe trata de un trastorno de herencia autosómica dominante121, producido por mutaciones que, al igual que en el síndrome AEC, afectan al dominio SAM, lo cual explica el considerable solapamiento clínico y molecular que existe entre estos 2 síndromes96,122. Para algunos autores ambos síndromes deberían ser considerados como un mismo trastorno121–127.
Síndrome trico-dento-óseoEs un trastorno de herencia autosómica dominante producido por diferentes mutaciones del gen DLX3, que se localiza en el cromosoma 17 (locus 17q21). Este gen, responsable también de la amelogénesis imperfecta, codifica la proteína DLX3128, que se expresa durante la embriogénesis y participa en la diferenciación de los derivados ectodérmicos, el tejido óseo y el tejido cartilaginoso129. Las manifestaciones clínicas son variables e incluyen alteraciones del esmalte dental, anomalías ungueales, pelo rubio y rizado, esclerosis y engrosamiento de los huesos del cráneo y alteraciones radiográficas como hipocalcificación y taurodontismo130.
Síndrome de WitkopTambién conocido como síndrome de dientes y uñas o disgenesia ungueal e hipodontia, este trastorno autosómico dominante se debe a una mutación en el gen MSX1, localizado en el cromosoma 4 (locus 4p16.1). El gen MSX1 interviene en la formación de determinados dientes (premolares, primeros molares y terceros molares) y de las uñas, en las que determina el grosor e integridad de la lámina ungueal131. Las características clínicas típicas son la displasia ungueal y la hipodontia, pero existe una considerable variabilidad en la expresión clínica. Aunque en algunos casos se han descrito alteraciones del cabello (fino o grueso), la mayoría de los individuos no presentan alteraciones en el cabello ni en la función de las glándulas sudoríparas132.
Síndrome Ellis-van CreveldEs un trastorno de herencia autosómica recesiva que se puede producir por mutación de los genes EVC o EVC2, siendo el fenotipo clínicamente indistinguible en cualquiera de los 2 casos133–135. Por otra parte, en aproximadamente el 30% de los casos con síndrome EVC no se encuentran mutaciones en ninguno de estos 2 genes, lo que sugiere que podría existir una mayor heterogeneidad genética136. Se caracteriza por alteraciones óseas, displasia ungueal, anomalías orofaciales y malformaciones cardiovasculares137. Los pacientes presentan talla baja, acortamiento acromesomélico de los miembros (más llamativo en el extremo distal) y pecho estrecho. Es frecuente encontrar polidactilia, sindactilia, genu valgo y diversas alteraciones dentales137,138.
Disóstosis acrodental de WeyersEsta entidad es un trastorno autosómico dominante cuyas mutaciones se localizan en los genes EVC y EVC2133,134,137. El patrón hereditario y la menor severidad fenotípica permiten distinguir este síndrome del EVC133. Su expresividad clínica es variable, caracterizándose por talla baja, hipotelorismo, orejas prominentes, polidactilia postaxial, anomalías orales (dientes irregulares, pequeños, con forma de clavija e hipodontia) y onicodistrofia (uñas displásicas o hipoplásicas)133,137.
Grupo 2Displasia ectódermica hidróticaLa DEHi o síndrome de Clouston se produce por mutaciones en el gen GJB6, localizado en el cromosoma 13 (locus 13q11-q12.1), que codifica la conexina 30139. Es un trastorno autosómico dominante particularmente común entre la población franco-canadiense140, y que solo excepcionalmente aparece de novo141. Las mutaciones en este gen también originan otros trastornos como la sordera neurosensorial autosómica dominante (DFNA3) y la autosómica recesiva (DFNB1)142.
Las conexinas son proteínas transmembrana que facilitan la comunicación intercelular140. Las moléculas de conexina forman hexámeros, llamados conexones, que interaccionan con otros conexones de células adyacentes para formar canales de unión intercelular o union gaps142. Estos canales permiten la difusión de pequeñas moléculas entre células y median el intercambio de señales y nutrientes, coordinando las actividades celulares y la respuesta a estímulos. En la piel se han identificado más de 10 conexinas diferentes. Cada conexina parece tener unas propiedades específicas, y su mutación produce también un trastorno cutáneo distinto. Así, la DEHi y el síndrome KID140 están provocadas por mutaciones en los genes GJB6 y GBJ2, que codifican las conexinas 30 y 26 respectivamente142. Estas 2 conexinas comparten el 76% de sus aminoácidos y se coexpresan en la capa córnea, las glándulas sudoríparas y los folículos pilosos140. Este solapamiento, a pesar de una diferente expresión tisular de las conexinas 26 y 30, implica algunas funciones comunes y quizá una interacción directa de ambas en muchos epitelios ectodérmicos. De hecho, ambas comparten características clínicas como la distrofia ungueal, la pérdida de pelo y la queratodermia plantar, y recientemente se ha descrito un paciente con una mutación en la conexina 30 cuyas manifestaciones clínicas eran similares a las del síndrome KID140.
Los 3 principales rasgos clínicos de la DEHi son la pérdida de pelo, la distrofia ungueal y la queratodermia palmoplantar140 (figs. 15 y 16). En contraste con la DEH, estos pacientes presentan tanto una dentadura como una función normal de las glándulas sudoríparas y sebáceas140,143. Las alteraciones del pelo se manifiestan como atriquia o hipotricosis que pueden empeorar progresivamente140; el pelo, fino y de crecimiento lento, presenta una estructura desorganizada y una birrefringencia disminuida144. Las mujeres presentan calvicie total, mientras que los varones presentan una expresión que varía desde un pelo rubio con áreas focales de alopecia hasta la alopecia total145. Las cejas y las pestañas son escasas o ausentes, así como el vello púbico y axilar140. Las alteraciones ungueales oscilan entre una apariencia casi normal y la micro o anoniquia; la lámina ungueal puede presentar engrosamiento, descamación, cambios de coloración, estriación y onicolisis140. Las alteraciones ungueales de estos pacientes pueden recordar a las de la paquioniquia congénita146. Algunos pacientes presentan también una queratodermia palmoplantar difusa y una discreta hiperpigmentación de la piel, particularmente evidente bajo el borde libre de las uñas y sobre las articulaciones digitales, las rodillas y los codos140,147. Se han descrito varios pacientes que presentaban siringofibroadenomas ecrinos148,149, y un caso aislado de pseudoainhum congénito150. Finalmente, el cuadro clínico puede acompañarse de estrabismo, conjuntivitis, pterigium, cataratas143, sordera neurosensorial, polidactilia y sindactilia147, pero no existe dismorfismo facial y el desarrollo físico general es normal.
.")
.")
Este trastorno de herencia autosómica recesiva se produce por mutaciones en el gen PVRL1 (poliovirus receptor-like 1), localizado en el cromosoma 11 (locus 11q23.2), que codifica a la proteína nectina-1151. Las nectinas son moléculas de adhesión celular calcio-independientes que actúan en las uniones intercelulares cooperando o no con las cadherinas. Hasta el momento se conocen 4 tipos diferentes de nectinas152. La nectina-1 se expresa en varios tejidos ectodérmicos, incluyendo la piel, los dientes y el pelo, fundamentalmente a nivel del estrato espinoso153. La mutación de este gen origina también la hendidura orofacial no sindrómica (OFC7)154. Los hallazgos clínicos característicos incluyen manifestaciones de displasia ectodérmica, labio/paladar hendido bilateral, retraso mental y sindactilia155. El cabello es escaso y corto, y a partir de la quinta década de la vida puede haber alopecia completa. Las cejas son escasas, especialmente en la zona lateral. También se observa xerosis cutánea, dermatoglifos hipoplásicos e hiperqueratosis palmoplantar progresiva. Entre las alteraciones dentales podemos encontrar retraso de la erupción dental, microdontia, hipodontia y anodontia. Las uñas son normales o levemente displásicas. Los pacientes presentan una facies peculiar, ovalada, con unas orejas de gran tamaño en anteversión. La sindactilia, que puede ser parcial, está frecuentemente presente en los dedos 2, 3 y 4, y en ocasiones es bilateral151. Otras manifestaciones incluyen sordera, anomalías genitourinarias o renales, anomalías del pezón y lordosis lumbar y variable retraso mental156. El diagnóstico diferencial debe establecerse principalmente con el síndrome EEC, con el que comparte la mayoría de las manifestaciones. La principal diferencia es el modo de herencia (el EEC es un trastorno autosómico dominante) y las malformaciones de los miembros, principalmente ectrodactilia, que aparecen en el 85% de los casos de EEC103,156.
En agosto de 2010 se describió una segunda nectinopatía debida a la alteración de la nectina 4, codificada en el gen PVRL4. Se trata de un síndrome similar al comentado en este apartado, conocido como Ectodermal dysplasia-Syndactyly Syndrome (EDSS), pero sin paladar hendido152.
Síndrome de displasia ectodérmica-fragilidad cutáneaEste trastorno autosómico recesivo, recientemente reclasificado dentro de las epidermólisis ampollosas simples, se debe a una mutación en el gen de la placofilina (PKP1), localizado en el cromosoma 1 (locus 1q32)157. La PKP1 es un componente estructural de los desmosomas que se expresa en los epitelios escamosos estratificados, el miocardio, las meninges y parte de los ganglios linfáticos158. Como el resto de las epidermólisis ampollosas se caracteriza por una gran fragilidad cutánea ante los traumatismos, eritema generalizado, alopecia, distrofia ungueal y queratodermia focal con dolorosas fisuras (fig. 17). Algunos pacientes presentan hipohidrosis, pero los dientes son normales en todos los casos157. El estudio histológico muestra espacios intercelulares ensanchados, separación de los queratinocitos, hendiduras intraepidérmicas, queratinocitos acantolíticos y diferentes grados de disqueratosis159, pero hasta la fecha no se ha descrito el desarrollo de carcinomas cutáneos.
Síndrome displasia ectodérmica-ectrodactilia-distrofia macular
El síndrome displasia ectodérmica-ectrodactilia-distrofia macular (DEEM) es un trastorno autosómico recesivo que se produce por una mutación del gen CDH3, localizado en el cromosoma 16 (locus 16q22.1) y que codifica la proteína cadherina-3160,161. Las cadherinas son moléculas de adhesión calcio-dependientes, con varios dominios extracelulares, una región transmembrana y otra intracelular. El extremo intracelular se une a la beta-catenina, implicada en la transcripción y adhesión celular. La expresión de CDH3 durante la embriogénesis desempeña un importante papel en el desarrollo normal. Se expresa al menos en la región orofacial y en los arcos faríngeos, así como en las extremidades, y podría ejercer un papel importante en la morfología de la mano humana161. Clínicamente, el síndrome DEEM se caracteriza por sindactilia, degeneración de la retina y cabello escaso160, aunque existe una significativa variabilidad clínica. Además de sindactilia, en algunos casos bilateral, puede haber ausencia de una o varias falanges o dedos totalmente hipoplásicos. Las manos suelen estar más afectadas que los pies161. Otro de los signos claves es la degeneración progresiva de la retina, con pérdida gradual de la visión. Se observa una prominente pigmentación en el polo posterior de la retina, junto con atrofia macular. Los pacientes muestran también hipotricosis con escasas cejas y pestañas y alteraciones dentales (hipodontia y dientes pequeños y muy separados)162.
Displasia odonto-onico-dérmicaEs un trastorno autosómico recesivo que se debe a mutaciones en el gen WNT10A, localizado en el cromosoma 2 (locus 2q35)163. Los genes WNT codifican una gran familia de glucoproteínas implicadas en una vía de señalización que es crucial para el desarrollo de los derivados ectodérmicos durante la embriogénesis y la vida adulta164,165. Así, WNT10A es importante para la formación de los dientes y los folículos pilosos, así como para la regeneración de la epidermis, las papilas linguales y la función de las glándulas sudoríparas164. Aunque la entidad se denomina displasia odonto-onico-dérmica debido a la afectación del pelo, sería más adecuado llamarla displasia trico-odonto-onico-dérmica163. Entre las lesiones cutáneas es característica la aparición en la región facial de unas placas eritematosas de aspecto reticulado, telangiectásico y atrófico, que se intensifican con el calor166,167. También es frecuente la hiperqueratosis palmoplantar, que se acompaña de hiperhidrosis y fisuración dolorosa168,169. Otras alteraciones incluyen la presencia de una lengua suave con papilas reducidas o ausentes, queratosis pilar170, hipotricosis, alteraciones dentales y alteraciones ungueales167. Algunos pacientes presentan un ligero retraso mental169. Diferentes mutaciones en este mismo gen son responsables de otra displasia ectodérmica de herencia autosómica recesiva, el síndrome de Schopf-Schulz-Passarge, con el que comparte ciertos rasgos clínicos (hipodontia, distrofia ungueal, queratodermia palmoplantar, lengua suave, hiperhidrosis e hipotricosis), pero estos pacientes además presentan otros rasgos como quistes en los párpados, y un riesgo aumentado para desarrollar tumores cutáneos165,171.
En conclusión, las displasias ectodérmicas son un grupo heterogéneo de trastornos hereditarios de difícil clasificación y con grandes similitudes clínicas. Los hallazgos biomoleculares de los últimos años nos han acercado a una clasificación clínico-funcional útil en la práctica clínica, pues relacionan las diferentes alteraciones genéticas en varias vías funcionales con un fenotipo particular. Sin embargo, dentro de las displasias ectodérmicas aún son muchos los trastornos cuya alteración genética no ha sido estudiada o identificada. No obstante, es importante que el dermatólogo conozca los principales síntomas y signos clínicos de estos trastornos para poder orientar el diagnóstico. Además, es importante identificar a las portadoras para poder ofrecerles consejo genético.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de la correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



