

Se han descrito diversas alteraciones moleculares en el melanoma. Los melanomas con mutaciones de BRAF suelen localizarse en zonas con exposición solar intermitente mientras que las alteraciones genéticas de KIT ocurren con mayor frecuencia en los melanomas acrales, mucosos y en los que se localizan en áreas con exposición solar crónica. Sorafenib, un inhibidor de BRAF, tiene un efecto citostático en la mayoría de los melanomas con mutaciones en la vía MAP cinasa, aunque en un pequeño subgrupo de estos melanomas es también capaz de promover la apoptosis. Imatinib, a través de la inhibición de KIT, posee un efecto citostático y citotóxico en aquellos melanomas con mutaciones de KIT, y probablemente en otro subgrupo de melanomas con otras alteraciones genéticas de KIT aún no perfectamente definidas. Para que estos tratamientos sean efectivos es imprescindible que se hayan seleccionado adecuadamente los pacientes, estableciéndose la existencia de alteraciones genéticas en la vía sobre la que se va a actuar.
A number of molecular alterations have been described for melanoma. Melanomas with BRAF mutations tend to be located in areas of intermittent sun exposure, whereas melanomas with KIT mutations mostly appear in acral areas, the mucosas, and areas of chronic sun exposure. Sorafenib, a BRAF inhibitor, has a cytostatic effect on most melanomas with mutations affecting the mitogen-activated protein kinase (MAPK) pathway, and is also capable of triggering apoptosis in a small subgroup of these melanomas. By inhibiting KIT, imatinib has a cytostatic and cytotoxic effect on melanomas with KIT mutations, and probably has the same effect on another subgroup of melanomas with other as yet imperfectly understood KIT mutations. For therapy to be effective, agents should be selected according to the pathways associated with the genetic mutations present in the melanoma.
El melanoma es un tumor maligno cuya incidencia ha aumentado constantemente en los últimos años. Aunque el diagnóstico en fases precoces ha supuesto una mejor supervivencia de los pacientes, la existencia de metástasis a distancia conlleva un mal pronóstico, con una mediana de supervivencia inferior a 1 año1. En las últimas décadas no se ha avanzado en el tratamiento del melanoma metastásico, de forma que la dacarbazina sigue siendo el quimioterápico más utilizado aunque pocos pacientes consiguen respuestas duraderas.
Los factores pronósticos más importantes en el melanoma cutáneo localizado son el espesor de Breslow y la ulceración2. Sin embargo, existen grandes diferencias en el comportamiento de pacientes cuyos melanomas son similares respecto a estos 2 factores. Esto ha llevado a considerar la existencia de diferencias genéticas, moleculares e inmunológicas significativas entre melanomas fenotípicamente superponibles. En los últimos años se ha producido un avance importante en el conocimiento del melanoma al identificarse diferentes vías moleculares que se encuentran alteradas. De crucial relevancia resulta el hecho de que ciertas alteraciones moleculares aparecen con más frecuencia en ciertas regiones anatómicas (mutaciones en c-KIT en melanomas mucosos y acrales) y en relación con una mayor o menor exposición al sol y según el tipo histológico (las alteraciones de BRAF son más frecuentes en melanomas de zonas no expuestas al sol y relacionadas con el tipo histológico de extensión superficial). Estos avances están abriendo nuevas posibilidades de tratamiento para el melanoma basadas en la modificación de las vías moleculares que se encuentran alteradas o implicadas en su desarrollo o en la respuesta inmunitaria del organismo frente a este tumor.
Tratamientos selectivos frente a dianas molecularesEl diagnóstico de un tumor se ha basado tradicionalmente en su aspecto histológico junto a la expresión de ciertos marcadores inmunohistoquímicos por los que las células tumorales reproducían las características del tejido y el grupo celular del que se originaban. Así sigue ocurriendo con el melanoma, en donde su conexión con la epidermis, su inicio en la capa basal y la agrupación en nidos de las células remedan lo que ocurre con los melanocitos y las proliferaciones benignas de éstos como son los nevus melanocíticos. Asimismo, la expresión de los mismos marcadores inmunohistoquímicos de los melanocitos, como la proteína S100 o el MelanA/MART1 ayudan a confirmar el diagnóstico.
Frente al rápido aumento en la incidencia del melanoma en los últimos años, la mortalidad debida a este tumor ha aumentado en mucha menor medida. La supervivencia media de los pacientes diagnosticados de melanoma ha mejorado considerablemente en las últimas 4 décadas (60% en 1960, 89% en 1990)3. El aumento en la incidencia no sólo se debe a un incremento real del número de melanomas sino también a los avances en el diagnóstico de este tumor, pero no es posible precisar el peso proporcional de cada uno de estos factores. Estos datos refuerzan la hipótesis de un grupo de investigadores de que existe un grupo de melanomas que, a pesar de ser localmente invasivos, crecen muy lentamente y no suelen comprometer la vida del paciente4. Lo que necesitamos en la actualidad de forma urgente son marcadores moleculares que permitan discriminar entre los melanomas con potencial agresivo, y que requerirían tratamientos adyuvantes tras cirugías extensas, de aquellos otros melanomas de buen pronóstico cuyo tratamiento se limitaría a la extirpación del tumor y revisiones periódicas.
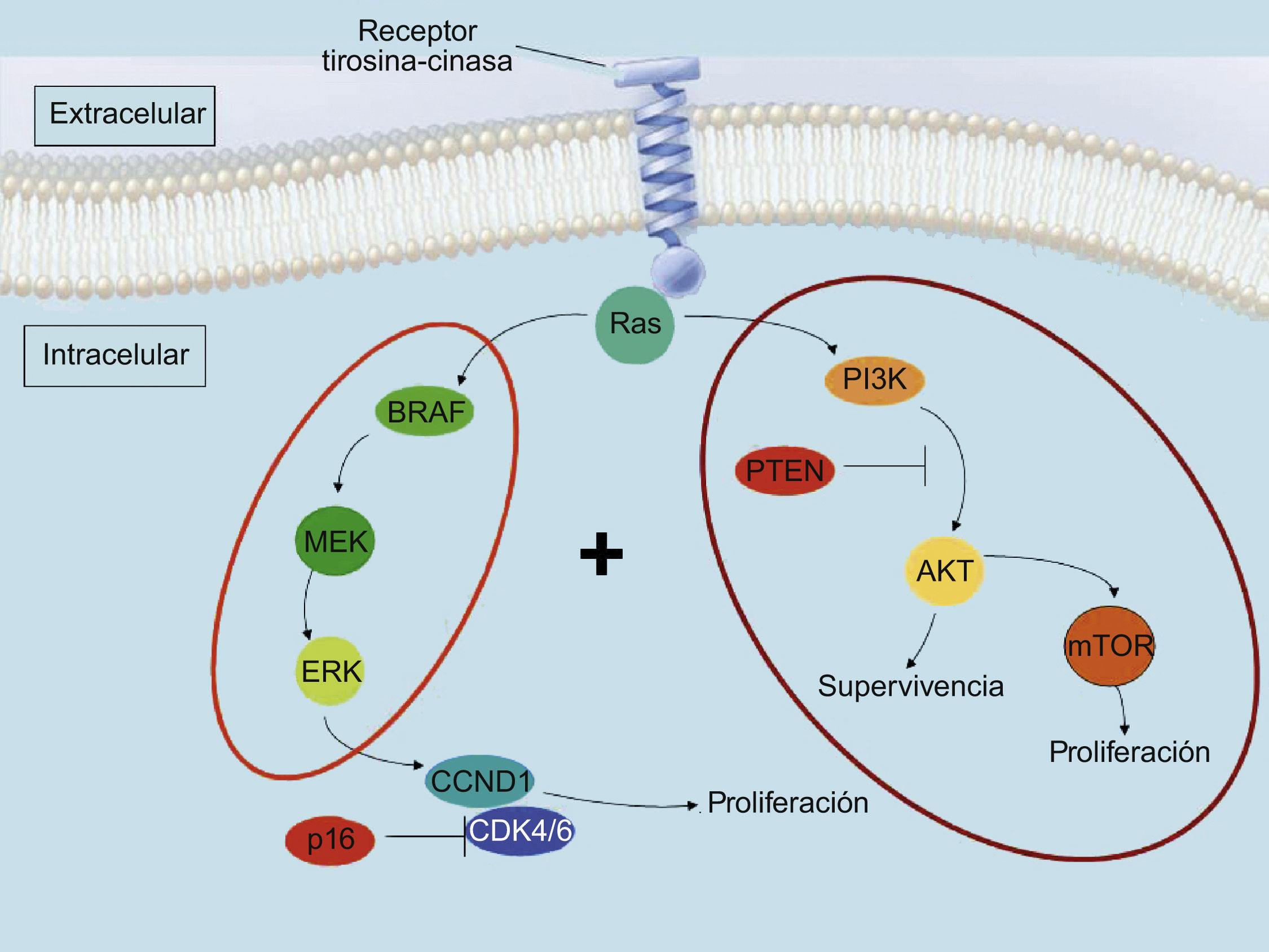
Vía de la MAPKEl desarrollo de un melanoma desde un melanocito sigue una serie de procesos en los que se acumulan alteraciones genéticas. Aunque esto no ocurre en todos los casos de forma sucesiva, los estadios progresivos se inician con el desarrollo de un nevus melanocítico, pasando por el nevus displásico, hasta llegar al melanoma in situ. A partir de aquí se desarrolla la fase de crecimiento radial del melanoma, y posteriormente la fase de crecimiento vertical. Llegado este punto, algunos melanomas metastatizan. Una de las alteraciones moleculares presentes en un mayor número de melanomas implica a la vía de señalización intracelular MAPK (mitogen-activated protein kinase), también conocida como ERK (extracellular-related kinase) (fig. 1). Esta vía de señalización intracelular incluye cuatro cinasas: RAS, RAF, MEK y ERK. En los melanomas, se han detectado mutaciones en la cinasa N-RAS en el 15% de los casos, mientras que B-RAF se encuentra mutada en el 50% de los casos (entre el 27 y el 70% según las series)5–7. Ambas mutaciones son excluyentes entre sí. Los nevus melanocíticos comparten con los melanomas una frecuencia similar de mutaciones en B-RAF8. La mutación más frecuente en B-RAF es la que ocurre en la posición 600 en donde el cambio de una timina por adenina lleva a la sustitución de valina por ácido glutámico. Este cambio origina una alteración en el propio dominio cinasa que conlleva una activación permanente de B-RAF y, por extensión, de la vía de la MAPK9.
 y fosfatidil-inositol 3 cinasa (PI3K), y de los puntos actuación de las proteínas supresoras tumorales CDKN2a y fosfatasa y tensina homólogo (PTEN).")
La mutación de cualquiera de estas cinasas causa la activación permanente de esta vía, lo que en principio debería determinar la proliferación continuada de las células. Sin embargo, la proliferación celular desencadenada por la mutación aislada en una de estas cinasas es controlada debido a un mecanismo de senescencia celular desencadenado al activarse una vía oncogénica. En concreto, la activación de la vía de la MAPK desencadenada por la mutación de BRAF, se controla en los melanocitos humanos aumentando la expresión del inhibidor de la cinasa 4A (INK4A), un inhibidor del ciclo celular10. Esto determina que en los nevus melanocíticos el crecimiento celular quede detenido. Para que se produzca una progresión hasta melanoma debe añadirse una mutación adicional en los genes supresores tumorales encargados, en condiciones normales, de detener la progresión iniciada por la mutación en la vía de la MAPK. En el caso de los melanomas familiares, en el 25–40% de los casos existe una mutación en el gen CDKN2A11. Este gen codifica 2 proteínas, p16INK4A y p14ARF, cuya función es detener la proliferación celular incontrolada. Respecto a los melanomas que ocurren fuera del contexto familiar, en el 25 al 50% de los casos existe una mutación en otro gen supresor tumoral, el homólogo de fosfatasa y tensina (PTEN)12,13. Por tanto, las mutaciones en la vía de la MAPK parecen ocupar un papel iniciador en el desarrollo de la modificación de los melanocitos, pero en la mayoría de los casos la puesta en marcha de mecanismos de senescencia celular aborta la progresión del fenotipo neoplásico, que concluye con la aparición de nevus melanocíticos.
BRAF no es un gen relacionado con una predisposición hereditaria para desarrollar cáncer. Los individuos con mutaciones de este gen en las células germinales no tienen una incidencia elevada de cáncer sino que desarrollan el síndrome cardio-facio-cutáneo14. Resulta especialmente interesante la relación de las mutaciones en BRAF con la exposición a la luz UV y, por extensión, con los diferentes tipos de melanoma. La mutación característica de BRAF en el melanoma, BRAFV600E, no suele ocurrir como consecuencia de la exposición a la luz UV, al menos en lo que se refiere a la exposición crónica a dicha luz. Las mutaciones en BRAF son más frecuentes en aquellos melanomas localizados en áreas del cuerpo sujetas a exposiciones intermitentes a la luz del sol, como el tronco y los brazos (59%), mientras que son más infrecuentes en los melanomas acrales (23%) y mucosos (11%), y se encuentran ausentes en los melanomas uveales15. Solo un 11% de los melanomas originados en zonas con exposición solar crónica tienen mutaciones en BRAF. Se ha identificado que las mutaciones de BRAF se asocian con un determinado fenotipo en los melanomas que las poseen frente a los que no tienen esta mutación, que consiste en aumento de la migración y coalescencia de las células en capas altas de la epidermis (crecimiento pagetoide), engrosamiento de la epidermis y delimitación neta de la piel circundante. Asimismo, las células de estos melanomas son más grandes, más redondeadas y pigmentadas16.
Pese a lo prometedor que parece en principio actuar farmacológicamente bloqueando la vía de la MAPK, varias evidencias experimentales han puesto en cuestión su utilidad. La dependencia de la vía de la MAPK observada in vitro en líneas celulares de melanoma y en modelos de xenotrasplante murino17,18, resultaba mitigada in vivo debido a la existencia de vías de activación alternativa de ERK mediante mecanismos autocrinos y paracrinos19. Por otra parte, las mutaciones en BRAF son fundamentales para iniciar el desarrollo del melanoma pero no son suficientes para justificar la transformación definitiva de los meloncitos y su mantenimiento20.
Dada la importancia que tiene la vía MAPK en el desarrollo del melanoma, se han desarrollado varias moléculas que inhiben selectivamente algunas de las cinasas implicadas. Sorafenib (BAY 43-9006) fue una de las primeras en desarrollarse9. Se trata de una bi-aril-urea que inhibe tanto a la cinasa BRAF normal como a la que posee la mutación BRAFV600E. Sorafenib posee actividad, además, frente a otras cinasas, como CRAF, VEGFR-3, VEGFR-2, PDGFR, c-KIT, y FLT3. Pese a los prometedores resultados in vitro para inhibir la vía de la MAPK, posteriormente se demostró tanto en líneas celulares de melanoma como en modelos de trasplante murino, que debido a sus características farmacológicas se precisaban concentraciones muy elevadas para conseguir una inhibición significativa de la vía MAPK5.
Sorafenib parece tener un mecanismo de acción antitumoral citostático. Por ello, la inhibición del crecimiento tumoral requiere la administración continuada del fármaco. Además, dentro de una toxicidad aceptable, se buscan las dosis mayores para tratar de inhibir la vía lo más completamente posible. Los principales efectos secundarios son astenia, anorexia, diarrea, exantema con descamación y síndrome pie-mano. Los estudios en fase I han establecido como dosis más apropiada 400mg, 2 veces al día21.
La Food and Drug Administration (FDA) ha aprobado la utilización de sorafenib en el tratamiento del carcinoma renal metastásico. En la patogenia de este tumor se encuentra claramente implicada la angiogénesis mientras que las mutaciones de BRAF no desempeñan ningún papel. Por ello, en la actualidad se ha afianzado la idea de que la actividad antitumoral de sorafenib se encuentra más relacionada con su actividad antiangiogénica derivada de la inhibición del VEGFR que del bloqueo de BRAF.
Los estudios clínicos en pacientes con melanoma metastásico utilizando sorafenib como monoterapia han ofrecido resultados desalentadores. En un estudio en fase I/II de 2005 que incluía a 22 pacientes con melanoma, sólo se consiguió una respuesta parcial en un paciente22. En otro estudio con 37 pacientes con melanoma metastásico, Eisen y sus colaboradores confirmaron que sorafenib posee una mínima actividad (1 respuesta parcial, 16% de pacientes con estabilización de su enfermedad) frente a este tumor en monoterapia23. Además, en este estudio se determinó si los melanomas tenían mutada la cinasa BRAF, condición que parecería necesaria para que sorafenib fuese efectivo si su efecto antimelanoma se encontraba mediado por la inhibición de esta molécula, y se halló que los efectos de sorafenib eran independientes del estado de esta cinasa. Los mejores resultados hallados hasta la fecha con sorafenib proceden de su uso combinado con dacarbazina. En un estudio aleatorizado y con doble enmascaramiento α, publicado en 2008, se comparó la actividad de la combinación de dacarbazina y sorafenib frente a dacarbazina y placebo en pacientes con melanoma en estadio III irresecable o con metástasis a distancia que no habían recibido antes otra quimioterapia24. Aunque la combinación de DTIC y sorafenib no consiguió una mejoría en la supervivencia global, sí se alcanzó una ventaja estadísticamente significativa en la supervivencia en ausencia de progresión de la enfermedad, y la toxicidad asociada fue llevadera. Sin embargo, no se determinó si los pacientes tenían mutaciones de BRAF como requisito previo a su inclusión en el estudio, por lo que existe la posibilidad de que la efectividad de sorafenib haya sido infraestimada.
En espera de otros fármacos que consigan un bloqueo más selectivo y completo de BRAF, se han desarrollado moléculas para bloquear la vía de la MAP cinasa. MEK es una cinasa que se encuentra justo por debajo de BRAF en la misma vía de señalización intracelular. Aunque no se han detectado mutaciones en MEK, el desarrollo de fármacos que bloquean esta cinasa ha demostrado utilidad para bloquear la activación desencadenada por las mutaciones en BRAF. De los fármacos desarrollados con este objetivo, 2 de ellos, PD0325901 y ARRY-142886, han mostrado unas características preclínicas (capacidad para inhibir en líneas celulares tumorales la actividad de la MAPK al disminuir la fosforilación de ERK, la última cinasa de esta vía; actividad antitumoral frente a un panel de xenoinjertos tumorales humanos; y unas propiedades farmacocinéticas y farmacodinámicas adecuadas) que han determinado su entrada en ensayos clínicos9.
Los últimos descubrimientos con sorafenib plantean un escenario discretamente diferente del esbozado aquí. Como ya hemos mencionado, un porcentaje muy elevado de melanomas tiene activada la vía de la MAPK. Esto puede ocurrir por mutaciones en NRAS o en BRAF. Existen ciertos melanomas en los que la señalización se debe a la activación de la cinasa CRAF a pesar de que no se conocen mutaciones en la propia cinasa. CRAF se encuentra al mismo nivel de BRAF, y en condiciones normales permanece desactivada y no participa en la activación de la vía MAPK. A diferencia de BRAF que solo activa la vía de la MAPK, CRAF activa también otras vías de proliferación celular (como MST-2, ASK-1, y NF-κB) y, además, al asociarse a las mitocondrias controla directamente la apoptosis, a través de la regulación de BAD y Bcl-225.
La mutación más frecuente en BRAF es la V600E, pero se conocen al menos otras 70 mutaciones en esta misma cinasa que conducen a una activación de BRAF de menor intensidad que la ocasionada por V600E, por lo que reciben el nombre de mutaciones de baja actividad26.
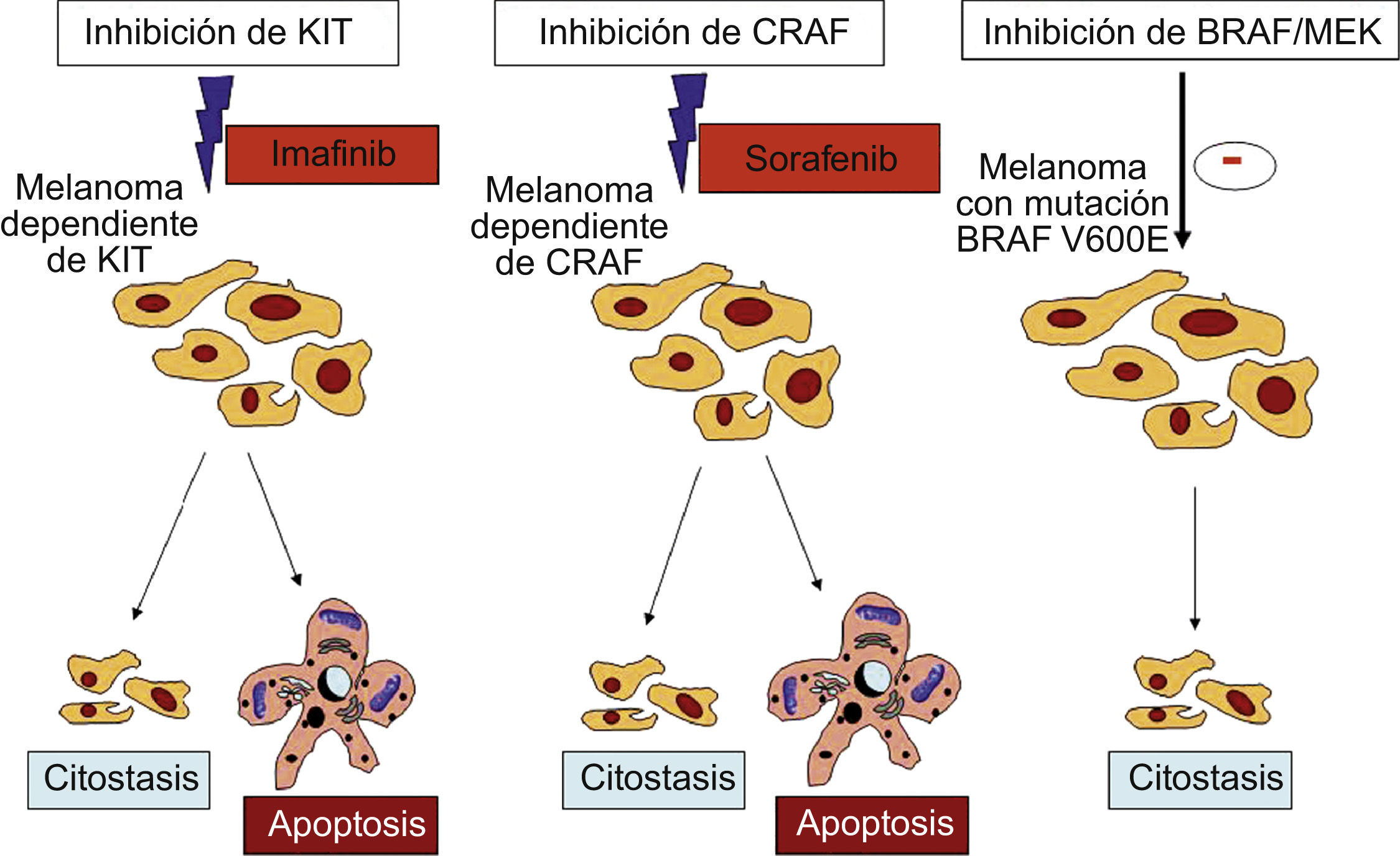
Los melanomas con la mutación V600E en BRAF necesitarían, como hemos dicho, de una mutación adicional en otras vías celulares, como PI3K o JAK/STAT. Estas vías se encuentran implicadas tanto en la proliferación celular como en la apoptosis. Por tanto, sorafenib, al detener solo la vía de la MAPK debido a la inhibición de V600E BRAF, tendría un efecto citostático reversible. Sin embargo, sorafenib inhibe de forma más selectiva a CRAF que a BRAF, por lo que podría ser muy efectivo en los 2 grupos de melanomas en los que la activación de la vía MAPK pasa a través de CRAF y no de BRAF: los que tienen una mutación en BRAF de baja actividad y los que tienen mutaciones en NRAS26,27. Aunque estos 2 grupos comprenden un porcentaje pequeño de melanomas, en ellos sorafenib tendría un efecto citostático y citotóxico debido a la implicación de CRAF en varias vías de proliferación celular y de apoptosis (fig. 2). Dicho efecto citotóxico resulta imprescindible para que el tratamiento sea efectivo. En líneas celulares de melanoma con las referidas mutaciones se ha demostrado que el efecto citotóxico se encuentra mediado a través de la inducción de la apoptosis debido a una disminución en la expresión de Bcl-225.
 (28).")
En los melanomas dependientes de KIT o de CRAF, imatinib y sorafenib, respectivamente, pueden resultar determinantes en el tratamiento del melanoma al inducir citostasis y apoptosis. Sin embargo, en los melanomas con la mutación BRAF V600E, sorafenib o los inhibidores de MEK solo consiguen una detención del ciclo celular (efecto citostático) (28).
Atendiendo a las alteraciones moleculares, el grupo de melanomas más numeroso es el que posee la mutación BRAF V600E. Desde un punto de vista genético, este grupo es además el más heterogéneo, ya que los melanomas aquí englobados probablemente suman a la mutación BRAF V600E otra alteración adicional, ya sea en PTEN, la ciclina D1, CDK2, CDK4, MITF, o AKT328. Esto explicaría la regresión incompleta observada con moléculas dirigidas a bloquear BRAF o MEK, debido a que estos tumores poseen vías alternativas compensadoras que les permite sobrellevar la inhibición de una sola de ellas. Por tanto, el futuro para un tratamiento efectivo de este grupo de melanomas recae en la utilización simultánea de fármacos dirigidos frente a las vías moleculares específicamente alteradas en estos tumores. En estudios preclínicos se ha demostrado que melanomas con la mutación BRAF V600E resistentes a la inhibición BRAF/MEK, responden cuando son sometidos a una inhibición dual de las vías MAPK/PI3K o MAPK/mTOR29–31. Si estos resultados se consolidan, el futuro para este grupo de melanomas pasa por el descubrimiento de fármacos altamente eficaces para bloquear cada una de estas rutas y, por supuesto, el éxito de estos tratamientos implica la necesidad de analizar de forma prospectiva las alteraciones genéticas de los tumores de los pacientes que van a recibir cada tratamiento.
Vía KITLa proteína KIT fue inicialmente descrita como una oncoproteína codificada por un retrovirus del sarcoma felino. El protooncogen KIT codifica un receptor tirosina-cinasa cuyo ligando es el stem cell factor (SCF), un factor de crecimiento fundamental en la hematopoyesis y en la formación de otros tipos de células, como los melanocitos y las células responsables de la motilidad intestinal. Las mutaciones en el receptor KIT causan su activación de forma permanente sin necesidad de unirse a su ligando. Se han identificado mutaciones en KIT en el tumor del estroma gastrointestinal (GIST), así como en ciertas leucemias, mastocitosis y seminomas. En el GIST de los seres humanos, las mutaciones en KIT suelen afectar a la porción yuxtamembrana del receptor. El dominio yuxtamembrana, tanto en KIT como en otros receptores tirosina-cinasa similares, tiene una función inhibidora sobre el propio receptor que se ve anulada cuando existen mutaciones que lo afectan. La importancia de reconocer los tumores en los que KIT está mutado deriva de la existencia de un fármaco, imatinib, capaz de inactivar este y otros receptores tirosina-cinasas.
La implicación de KIT en el melanoma ha estado sujeta a controversia y todavía no se conoce con exactitud. Dos importantes trabajos publicados en 2005 y 2006 encontraron que imatinib era inefectivo en el tratamiento del melanoma con independencia de que el tumor expresara o no el receptor KIT en su superficie32,33. Sin embargo, con posterioridad se ha analizado de forma más precisa el estado de este receptor en los diferentes tipos de melanoma. Curtin y colaboradores encontraron mutaciones o aumentos en el número de copias del gen KIT en los melanomas mucosos, en los acrales y en menor medida también en los localizados en piel con exposición solar crónica34. Sin embargo, los melanomas localizados en áreas con exposición solar intermitente tenían una frecuencia muy baja de mutaciones en KIT. Estos resultados ofrecen un panorama según el cual los melanomas caracterizados por un patrón de crecimiento lentiginoso, en los que las células de melanoma se disponen de forma aislada en la capa basal antes de invadir la dermis (como suele ocurrir en aquellos localizados en mucosas, zonas acrales y en piel con exposición solar crónica), suelen tener mutaciones en KIT. En contraposición, los melanomas con un patrón de crecimiento pagetoide, con células que ascienden en la epidermis y confluyen formando grupos o nidos, característico de los melanomas de extensión superficial que suelen localizarse en piel con exposición solar intermitente, tienen con mayor frecuencia mutaciones de BRAF.
La relevancia del tratamiento con imatinib en el melanoma ha quedado establecida tras la publicación en 2008 de los casos de 2 pacientes con melanoma mucoso metastásico con mutaciones de KIT que tuvieron respuestas dramáticas a este tratamiento35,36. La dependencia del tumor frente a imatinib se evidenció por el hecho de que uno de los pacientes requirió una disminución temporal de la dosis de imatinib que se acompañó de una reaparición de las metástasis que, sin embargo, volvieron a responder a un aumento de la dosis de imatinib.
Parece claro que la mera presencia del receptor en la superficie del melanoma no anticipa su respuesta al tratamiento con imatinib. Los candidatos ideales para este tratamiento serían aquellos pacientes con melanomas en los que KIT se encuentra fosforilado y activa a su vez vías celulares esenciales para la supervivencia y crecimiento28. Se ha demostrado que el tratamiento con imatinib de líneas celulares de melanoma mucoso con mutaciones activadoras de KIT consigue una inhibición simultánea de las vías MAPK, PI3K/AKT y JAK/STAT37. La supresión combinada de estas tres vías supone una detención del ciclo celular a la vez que se induce la apoptosis debido a la reducción de la expresión de Bcl-2 y survivina. Los máximos beneficios clínicos deben esperarse en aquellos melanomas en los que la inhibición de KIT tenga efectos citotóxicos al desencadenar la apoptosis (fig. 2).
En la actualidad, se conocen al menos tres alteraciones moleculares que explicarían una función exacerbada de la vía KIT. Dos de ellas ya han sido mencionadas, la existencia de mutaciones activadoras y el aumento en el número de copias del gen KIT, y la tercera sería la presencia de una activación constitucional de la vía CDK4. Referente a esta última, se ha demostrado la existencia de un grupo de melanomas sin mutaciones en la vía de la MAP cinasa (BRAF y NRAS normales) que sobreexpresan tanto la vía KIT como la CDK438. A pesar de no tener mutaciones en KIT, estos melanomas demostraron una alta expresión del receptor KIT fosforilado, subrayando que este receptor se encontraba activado. En experimentos in vitro y en modelos de melanoma in vivo, los melanomas con sobreexpresión de estas 2 vías no respondieron al tratamiento con inhibidores de BRAF, sin embargo, su crecimiento resultó inhibido con imatinib.
Respecto a la relación entre la expresión de KIT y la existencia de mutaciones, recientemente se han publicado trabajos que demuestran que los melanomas con mutaciones de KIT suelen tener una expresión elevada (más del 50% de las células) de este receptor en la inmunohistoquimia39. Expuesto de una forma más práctica, aunque sólo una proporción de los melanomas con expresión elevada de KIT en la inmunohistoquimia tendrán mutaciones, no debemos esperar la existencia de mutaciones en los melanomas con una baja expresión de KIT (menos del 10% de las células).
Recientemente, Wu y colaboradores han encontrado la existencia de una asociación estadísticamente significativa entre la presencia de hiperpigmentación y la expresión de KIT mediante inmunohistoquimia en los melanomas40. Estos autores estudiaron 70 melanomas, localizados en diferentes zonas, y con tipo histológico tanto epitelioide como fusiforme. Curiosamente, solo 2 de los 70 tenían una mutación de KIT por lo que no pudieron establecer relaciones en este sentido. La correlación de hiperpigmentación y aumento de la expresión de KIT es comprensible si consideramos que KIT se encuentra implicado en la melanogénesis. Este papel ha quedado refrendado al encontrar una mutación en el ligando de KIT, con resultado de un aumento de su función, en pacientes con hiperpigmentación progresiva familiar41. Este síndrome hereditario autosómico dominante se caracteriza por manchas cutáneas hiperpigmentadas presentes desde los primeros meses de vida y que aumentan en número y tamaño con la edad.
En algún caso se han comunicado respuestas al tratamiento con imatinib de pacientes con melanomas diseminados en ausencia de mutaciones relevantes de KIT. En concreto, Kim y sus colaboradores publicaron un estudio en el que, de los 21 pacientes con melanoma metastásico tratados con imatinib, aquel cuyas células tumorales mostraron la expresión más intensa de KIT con inmunistoquimia, consiguió una respuesta completa42. Los melanomas incluidos en este estudio no tenían mutaciones en los exones 9 ni 11 (los más frecuentemente mutados en los tumores del estroma gastrointestinal que responden a imatinib), ni en los exones 13, 15 o 17. El melanoma del paciente que respondió tenía un procesamiento postranscripcional alternativo (alternative splicing) en el exón 15 que, sin embargo era compartido por otros 4 melanomas de pacientes que no respondieron a imatinib.
Existen múltiples interrogantes acerca de las anomalías de la vía KIT en el melanoma y de la respuesta a imatinib. ¿Responden de manera diferente los melanomas con mutaciones que los que tienen aumento en el número de copias del gen o los que tienen sobreexpresión de las vías KIT y CDK4? ¿Existe un grupo de melanomas con sobreexpresión de KIT demostrable mediante inmunohistoquimia, en los que KIT resulta fosforilado y por tanto la vía activada, en ausencia de las tres alteraciones moleculares mencionadas? En este sentido, podríamos extrapolar a algunos melanomas cutáneos lo que ha sido demostrado en los melanomas uveales, en los que existe un aumento en la expresión de KIT y KIT fosforilado, cuyo crecimiento resulta inhibido con imatinib, pero la hiperfunción de esta vía no se debe a la presencia de mutaciones sino a la liberación autocrina y paracrina por el melanoma del ligando de KIT, el stem cell factor43.
El éxito de una terapia como ésta vuelve a subrayar la necesidad de seleccionar adecuadamente a los candidatos para recibirla (melanomas con alteraciones del gen KIT), ya que, de no hacerlo así, se corre el riesgo de desestimar tratamientos cuya utilidad nunca alcanzará diferencias estadísticamente significativas en un grupo de melanomas no seleccionado44.
ConclusionesLa biología molecular nos está permitiendo diseccionar el melanoma, de forma que los diferentes subtipos clinicopatológicos se están reagrupando según las alteraciones moleculares que presentan. Una de las conclusiones más sorprendentes de los trabajos referidos es la correlación mencionada entre patrón de crecimiento pagetoide o lentiginoso con mutaciones respectivamente en BRAF o KIT. A pesar de que sorafenib e imatinib se postulan como tratamientos para un subgrupo de melanomas, el futuro parece decantarse por tratamientos combinados con diferentes fármacos que a su vez interfieran con las diferentes vías moleculares precisas para el mantenimiento del melanoma.



