






Introducción
El síndrome antifosfolípido (SAF) es una trombofilia autoinmune adquirida que se caracteriza por la presencia de anticuerpos antifosfolípido (AcAF), fenómenos trombóticos y/o pérdidas fetales recurrentes. Aún existe controversia en cuanto a su diagnóstico y manejo terapéutico. Vamos a comentar el significado de los diferentes AcAF, intentar exponer de forma clara y concisa los criterios clínicos y analíticos que definen el cuadro, sus variadas manifestaciones clínicas, con especial mención a las lesiones cutáneas, y finalmente expondremos las pautas que se manejan hoy día en el tratamiento y prevención de los fenómenos trombóticos y la morbilidad obstétrica.
Los anticuerpos antifosfolípido
Los AcAF son un grupo heterogéneo de inmunoglobulinas (Ig), fundamentalmente IgG, IgM e IgA, que se ligan al complejo formado por fosfolípidos aniónicos, principalmente cardiolipina, y proteínas plasmáticas que se ligan a estos fosfolípidos. Estas proteínas son fundamentalmente beta 2-glucoproteína I (B2-GP1) y protrombina, aunque existen otros anticuerpos (Ac) que van dirigidos contra proteína C, proteína S, anexina y antitrombina III, entre otros. Estos anticuerpos provocan la aparición de trombosis alterando la función que estas proteínas tienen en el proceso de la coagulación, además de actuar directamente sobre el endotelio vascular y el sistema inmune 1. Pueden aparecer Ac capaces de ligarse directamente a fosfolípidos sin la presencia de proteínas plasmáticas. Esto suele ocurrir en situaciones como infecciones, ingesta de fármacos o neoplasias, y no se asocian con fenómenos trombóticos.
Se detectan en el laboratorio de dos formas: a) por su interferencia con las pruebas de coagulación fosfolípido-dependientes: anticoagulante lúpico (aCL), o b) mediante ELISA. Mediante esta segunda forma podemos detectar Ac dirigidos contra el complejo fosfolípido-proteína plasmática utilizando el fosfolípido como antígeno (fundamentalmente cardiolipina): anticuerpos anti-cardiolipina (aCA), o utilizando directamente extractos purificados de proteína como antígeno (fundamentalmente la B2-GP1): anticuerpos anti beta 2-glucoproteína1 (anti B2-GP1) (tabla 1).

Anticoagulante lúpico
Su determinación debe realizarse siguiendo los criterios dictados por la sociedad internacional de trombosis y hemostasis 2. Esta prueba detecta tanto Ac contra B2-GP1 como protrombina. En pacientes con enfermedad autoinmune los asociados a anti B2-GP1 parecen correlacionarse mucho mejor con trombosis. Se están estudiando métodos para poder discriminar entre ambos Ac y confirmar este dato 3. Es más específico pero menos sensible que los aCA, y su correlación con el riesgo de trombosis y morbilidad obstétrica es muy alta, sobre todo en pacientes con lupus eritematoso sistémico (LES) 3,4. En sujetos en tratamiento con anticoagulantes orales su determinación puede ser imposible.
Anticuerpos anti-cardiolipina
Detecta tanto Ac ligados a complejos fosfolípido-proteína, como Ac que se ligan directamente a fosfolípidos. Es una prueba más sensible, pero menos específica que el aCL. En la reunión de Sydney se definió la cifra de 40 GPL o MPL como el límite entre títulos bajos, que no serían significativos, y medios/altos 3. Suelen determinarse los IgG e IgM que son los incluidos como criterios diagnósticos. En presencia de crioglobulinas y factor reumatoide pueden existir falsos positivos para aCA IgM a títulos bajos 3. También se pueden detectar aCA IgM de forma transitoria y a títulos bajos en pacientes con procesos infecciosos, neoplasias e ingesta de algunos fármacos sin asociarse a fenómenos trombóticos. Estos Ac suelen ligarse directamente a fosfolípidos sin la presencia de proteínas plasmáticas, y por ahora no hay técnicas habituales de laboratorio para poder diferenciarlos de otros Ac que sí se asociarían a trombosis. Los aCA IgA parecen ser más frecuentes en un subgrupo de pacientes con enfermedad autoinmune, trombocitopenia, úlceras cutáneas y vasculitis, pero no se consideran criterio diagnóstico 3.
Anticuerpos anti B2-glucoproteína 1
Suelen detectarse junto a otros AcAF, aunque pueden aparecer aislados en el 3-10 % de los SAF. Tienen buen valor predictivo, incluso mejor que los aCA, del riesgo de trombosis y patología obstétrica sobre todo a títulos altos 5,6. Todavía se está estandarizando la titulación de sus niveles y por ahora se recomienda considerar positivos los superiores al percentil 99 de los controles 3. En el laboratorio se detectan los Ac IgG e IgM. Los IgA no tienen aún bien establecida su utilidad clínica, y por ahora no se aconseja su determinación 3. En pacientes con factor reumatoide positivo o crioglobulinas pueden existir falsos positivos, sobre todo para Ac IgM.
El significado de otros Ac dirigidos contra proteínas diferentes de la B2-GP1 o antifosfolípidos diferentes de la cardiolipina no está claro, ni sus técnicas de detección en el laboratorio están bien estandarizadas por el momento. Se están investigando de forma especial Ac contra protrombina, complejo protrombina-fosfatidilserina y anti-fosfatidil-etanol-amina, pero aún no hay datos concluyentes para recomendar su determinación 3,6.
Criterios diagnósticos del síndrome antifosfolípido
El SAF es una enfermedad autoinmune, multisistémica, con etiología multifactorial genética y ambiental, que se caracteriza por la presencia de AcAF, trombosis (arterial o venosa) y/o pérdidas fetales de repetición. Para establecer un diagnóstico definitivo se definieron unos criterios en el Octavo Congreso Internacional sobre anticuerpos antifosfolípido de Sapporo 1998 7, que fueron revisados en el Decimoprimer Congreso de Sydney 2004 3 (tabla 2).

El hecho de que existan riesgos añadidos de trombosis hereditarios o adquiridos no es razón para excluir el diagnóstico de SAF. De hecho, se recomienda que se reconozcan dos grupos de pacientes: a) con factores de riesgo trombótico asociados y b) sin factores de riesgo trombótico 3. Los principales factores de riesgo trombótico a tener en cuenta son: edad (superior a 55 años en hombres y 65 en mujeres), factores de riesgo de enfermedad cardiovascular (hipertensión arterial, diabetes, hiperlipidemia y obesidad), síndrome nefrótico, tabaquismo, anticonceptivos orales, trombofilias hereditarias, neoplasias, inmovilización y cirugía. Es importante tenerlos en cuenta a la hora del diagnóstico para establecer medidas preventivas 3.
Manifestaciones clínicas asociadas al síndrome antifosfolípido
Se trata de manifestaciones clínicas muy frecuentes en el SAF y que fueron definidas en la reunión de Sydney 3. No se pueden utilizar para establecer un diagnóstico definitivo de SAF, pero en enfermos con criterios analíticos que no cumplan los clínicos, su presencia nos debe hacer pensar en un «probable SAF» (tabla 3).

Clínica del síndrome antifosfolípido
La clínica del SAF puede ser muy variada, dado que las venas y arterias de cualquier órgano pueden verse afectadas por los fenómenos trombóticos. La manifestación más común en el sistema venoso es la trombosis a nivel profundo en extremidades inferiores, con o sin tromboembolismo pulmonar, y a nivel arterial el accidente cerebrovascular agudo (ACVA), pero puede verse envuelto cualquier otro sistema venoso o arterial incluyendo el superficial, miocárdico, retiniano e intra-abdominal. La afectación a nivel placentario condicionaría la patología obstétrica.
En la tabla 4 se recogen las manifestaciones clínicas más frecuentes de una serie de 1.000 pacientes diagnosticados de SAF siguiendo los criterios de la reunión de Sapporo, recogidos por el grupo europeo para el estudio del SAF 8.

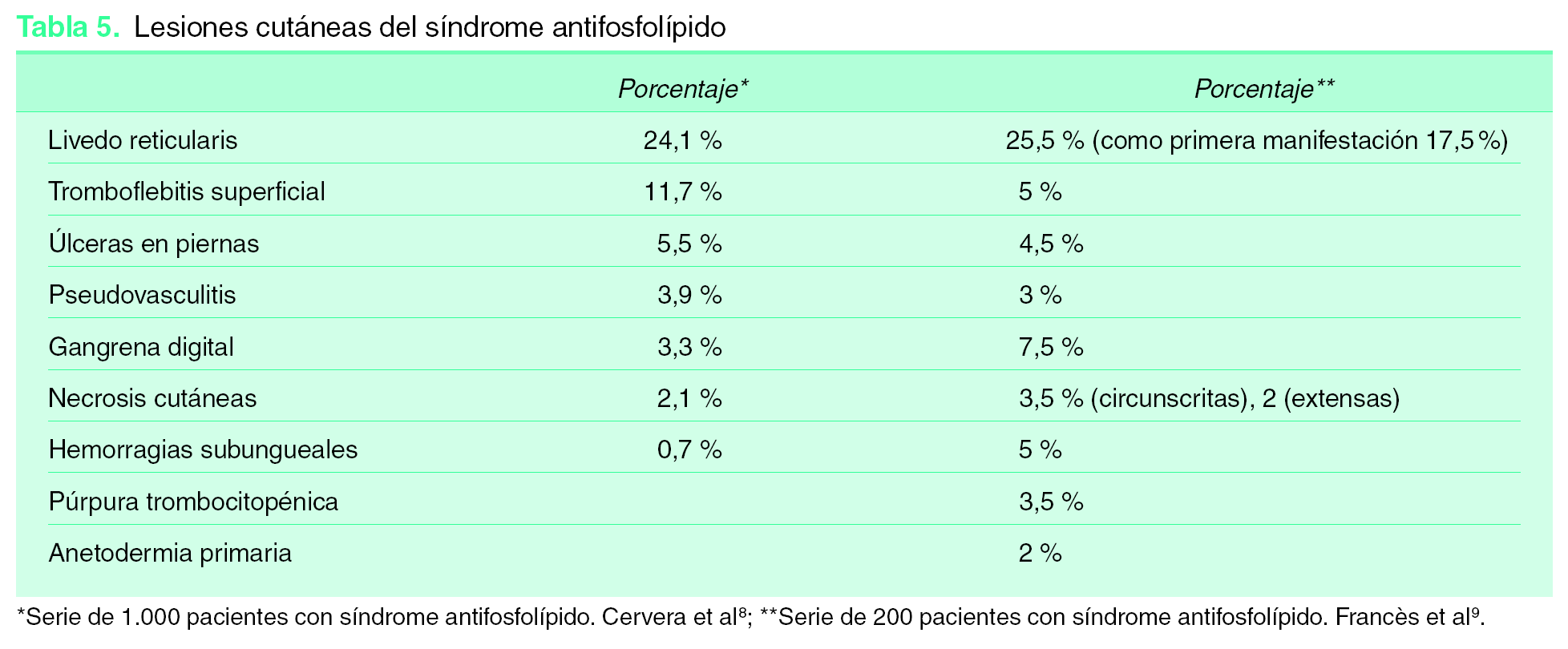
Afectación cutánea en el síndrome antifosfolípido
Existen lesiones muy frecuentes, otras lo son menos, pero todas ellas nos pueden ayudar en el momento del diagnóstico. En ocasiones pueden ser el primer o el único síntoma de la enfermedad 9 (tabla 5):
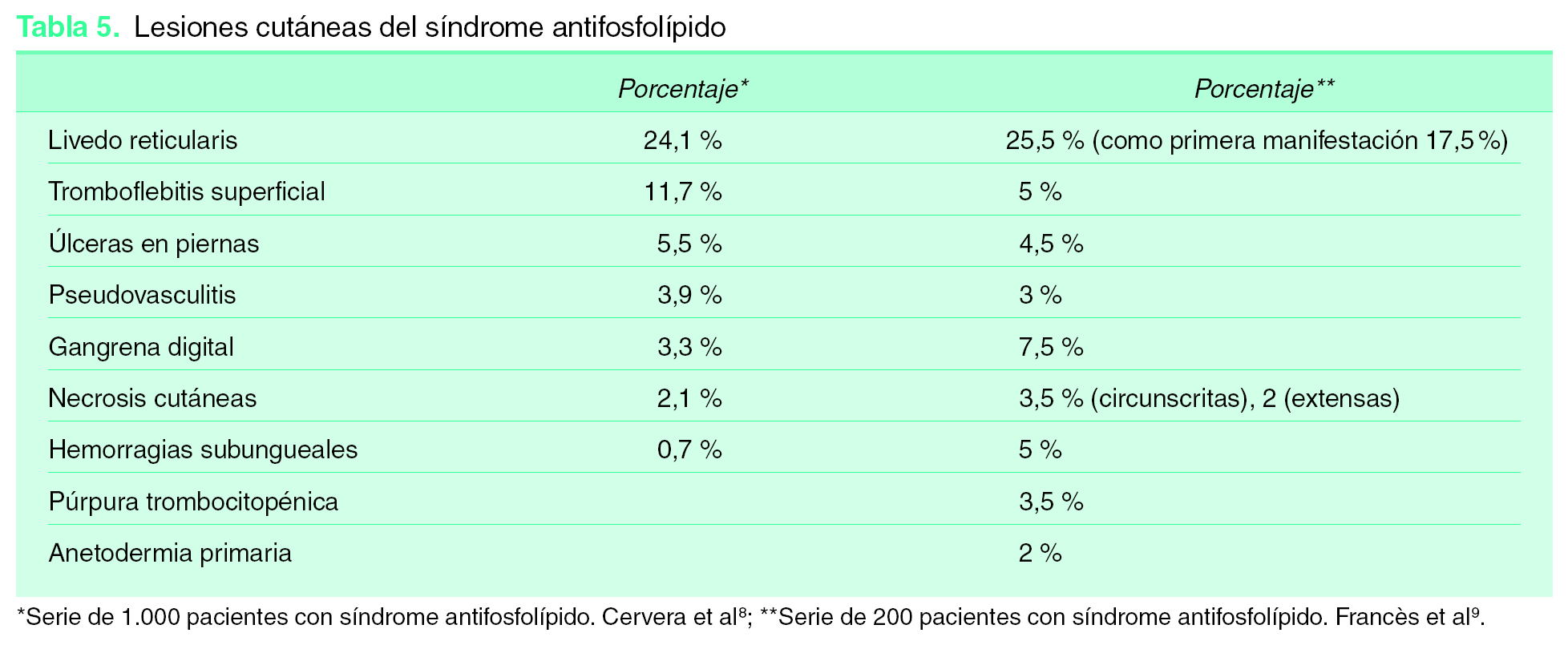
1. Livedo reticularis (LR): muy frecuente. Puede aparecer hasta en el 25 % de los SAF y en un 20,4 % como primera manifestación 8. Ha sido incluida entre las manifestaciones clínicas asociadas a SAF 3. Es extensa, afectando a las extremidades, el tronco y las nalgas, aunque al inicio pueden afectarse sólo el dorso de manos y pies. Su patrón es fino, reticulado e irregular. Su presencia se asocia con mayor frecuencia a trombosis arteriales con sintomatología cerebral y ocular, valvulopatía cardiaca, hipertensión arterial y fenómeno de Raynaud. Es menos frecuente entre los pacientes sólo con trombosis venosas.
2. Tromboflebitis superficiales.
3. Úlceras en piernas: úlceras post-flebíticas, ulceraciones secundarias a necrosis cutáneas, lesiones tipo atrofia-blanca o livedo vasculitis.
4. Grandes lesiones ulcerosas tipo pioderma gangrenoso.
5. Hemorragias subungueales.
6. Lesiones purpúricas.
7. Pseudovasculitis: púrpura, pequeñas pápulas, nódulos eritemato-violáceos y lesiones necróticas; es preciso biopsiar estas lesiones para diferenciarlas de verdaderas vasculitis.
8. Necrosis cutáneas extensas. Habitualmente de aparición brusca, con placas necróticas con borde activo purpúrico y lesiones ampollosas.
9. Gangrenas digitales.
10. Anetodermia primaria.
A nivel histológico aparece una trombosis de venas, arteriolas y arterias de pequeño-mediano tamaño en dermis o tejido celular subcutáneo, sin componente inflamatorio en la pared vascular. En la LR es difícil encontrar estas alteraciones (a excepción de la LR que aparece en el síndrome antifosfolípido catastrófico), y cuando aparecen lo hacen tanto en el centro como en el borde de la lesión 9. La histología de la anetodermia es similar a la que aparece en otras entidades, pero en muy raras ocasiones también se puede encontrar trombosis.
Síndrome antifosfolípido primario y secundario
La enfermedad puede aparecer de forma aislada, hecho que ha venido denominándose «síndrome antifosfolípido primario» (SAFP), o asociada a otras enfermedades, fundamentalmente autoinmunes: «síndrome antifosfolípido secundario» (SAFS). En una serie de 1.000 pacientes diagnosticados de SAF8 un 53 % fueron SAFP y el resto SAFS asociados a otras patologías. Entre ellas, la más frecuente fue el LES o cuadros tipo lupus que correspondían a un 41 % de los casos. El resto de asociaciones fueron síndrome de Sjogren primario (2,2 %), artritis reumatoide (1,8 %), esclerodermia sistémica (0,7 %), vasculitis sistémicas (0,7 %) y dermatomiositis (0,5 %). No se han encontrado diferencias significativas entre las manifestaciones clínicas o serológicas del SAFP y el SAFS8-10, y no se aconseja el uso de esta terminología3.
Dada su frecuente asociación, siempre que se diagnostique un SAF es preciso descartar la existencia de algún proceso autoinmune, sobre todo LES. La aparición del cuadro asociado puede ocurrir a lo largo de la evolución del proceso. En una serie de 128 pacientes diagnosticados de SAFP, seguidos durante 14 años y con una media de duración de la enfermedad de 8,2 años, sólo en un 8 % apareció LES, y en un 5 % cuadros tipo lupus11.
De forma inversa, cuando tengamos un paciente con LES, siempre hemos de tener presente la posibilidad del desarrollo de un SAF a lo largo de su evolución. Los AcAF son muy frecuentes en pacientes con LES. Entre ellos, el 15-30 % tiene aCL, y un 86 % aCA12. Tras un seguimiento de pacientes con LES y AcAF, en 7 años un 30 % había desarrollado SAF, y en 20 años la cifra aumentó a 50-70 %13. Los pacientes con LES que acaban desarrollando SAF suelen tener con mayor frecuencia artritis, livedo reticularis, trombocitopenia, leucopenia, disminución de C4, anemia hemolítica autoinmune y valvulopatía cardiaca3.
Síndrome antifosfolípido catastrófico
Se trata de una forma clínica peculiar de SAF que se define por la presencia de trombosis simultánea en tres o más órganos. Es rápidamente progresiva, con una mortalidad que puede llegar hasta el 50 %. Aunque pueden afectarse grandes vasos, es típica una microangiopatía trombótica multiorgánica de vasos de pequeño calibre que debe evidenciarse histológicamente. La afectación renal es la más frecuente (78 %), seguida de la pulmonar (66 %), cerebral (60 %), cardiaca (50 %), dermatológica 50 % (livedo reticularis, necrosis cutáneas extensas y gangrenas digitales), hepática 40 %, suprarrenal 30 %, y con menor frecuencia afectación gastrointestinal, esplénica, de la vesícula biliar, médula ósea o aparato reproductivo. También son frecuentes la trombocitopenia (60 %), anemia hemolítica (39 %) y coagulación intravascular diseminada (CID) 19 % 14,15. Es un cuadro infrecuente (0,8 % de los SAF en una serie de 1.000 pacientes) 8. Hasta en el 65 % de los casos se detecta un factor precipitante como infecciones, cirugía, traumatismos, neoplasias, inicio de anticoagulación, brotes de LES, ingesta de fármacos (anticonceptivos, tiacidas, captopril, danazol, etc.), inducción de ovulación y vacunaciones. Puede aparecer tanto en el SAFP (49 %) como en el SAFS, fundamentalmente asociados a LES o tipo lupuslike (45 %)14,15. El diagnóstico diferencial puede llegar a ser difícil, debiéndose realizar con cuadros sépticos, púrpura tombocitopénica trombótica, síndrome hemolítico-urémico y CID.
Manejo de los pacientes con anticuerpos antifosfolípido y síndrome antifosfolípido
En los pacientes con un cuadro trombótico establecido, el pilar del tratamiento es la anticoagulación oral (AO). Sólo existe alguna discrepancia en torno a los niveles de la misma, y si debe ser o no definida en función del territorio afectado, si es un primer episodio, o es recurrente. La mayor controversia surge en torno al tratamiento preventivo en los pacientes con AcAF que aún no han desarrollado sintomatología clínica y las pautas durante el embarazo (tabla 6).

Pacientes con anticuerpos antifosfolípido, sin manifestaciones clínicas de síndrome antifosfolípido
En un paciente totalmente asintomático, en el cual se ha detectado la presencia de AcAF en una analítica de rutina, por ejemplo un preoperatorio, y que mantiene sus títulos positivos en posteriores determinaciones (debe repetirse la titulación con intervalo de al menos 12 semanas): si el aCL es negativo y sólo son positivos los aCA a títulos < 40, no se aconseja tomar ninguna medida, pero si es positivo el aCL o los títulos de aCA son > 40, hay que intentar controlar otros posibles riesgos trombóticos asociados (tabaco, obesidad, hiperlipidemia, hipertensión arterial e ingesta de anticonceptivos), y cuando se produzca una situación de especial riesgo, como intervenciones quirúrgicas o inmovilizaciones prolongadas, se consideraría la utilización de heparina 16. La utilización de ácido acetilsalicílico (AAS) de forma profiláctica a dosis bajas (75-100 mg/día) es aconsejada por la mayoría de los autores 16,17, aunque se están realizando estudios para consensuar esta actuación e incluso considerar la posible asociación de AO con unos niveles del cociente internacional normalizado (INR) > 1,5 18.
La presencia de AcAF en pacientes con LES, sin manifestaciones clínicas de SAF, obliga a seguir el mismo criterio que en el apartado anterior (aCL negativo y aCA IgG positivos < 40 no hacer nada). Si hay aCL positivo o los aCA IgG son positivos a título > 40, además del AAS se aconseja la administración de hidroxicloroquina 16,19.
Primer episodio trombótico en pacientes con anticuerpos antifosfolípido
1. Venoso. Anticoagulación: iniciar con heparina durante 5 días, solapando con el inicio de AO a INR 2-3. La mayoría de los expertos recomiendan que ésta sea indefinida 20, pero no está tan claro cuando el primer episodio aparece asociado a otros factores de riesgo trombótico importantes que pueden ser controlados (por ejemplo el uso de anticonceptivos orales, embarazo y cirugía) 16.
2. Arterial cerebral. AO indefinida a INR 2-3 21,22, aunque algunos autores no están de acuerdo y proponen que el AAS 325 mg/día sería tan eficaz como la AO y con menos efectos secundarios 20, sobre todo en pacientes con títulos bajos o moderados de aCA y ausencia de otros factores de riesgo trombótico 12.
3. Arterial no cerebral. No existen muchos estudios en este tipo de pacientes pero la mayoría recomienda AO a INR 2-3 indefinido, aunque depende de la localización y severidad del cuadro 12,20,21.
Pacientes con trombosis recurrentes a pesar del tratamiento
No hay aún pautas bien establecidas aunque la mayoría recomienda AO a INR 3-4 o heparina 20,21. Algunos recomiendan asociar antiagregantes plaquetarios 20 o AAS en caso de trombosis cerebral recurrente 12. El uso de corticoides o inmunosupresores sólo estaría justificado en casos de episodios repetidos de trombosis 16.
Síndrome antifosfolípido catastrófico
El tratamiento no está bien definido y continúa siendo insatisfactorio. Además de AO se recomiendan altas dosis de esteroides por vía intravenosa, plasmaféresis, inmunoglobulinas intravenosas y antibióticos si existe o se sospecha infección. Algunos casos con trombocitopenias severas han sido tratados con éxito con rituximab 15,16.
Lesiones cutáneas en el síndrome antifosfolípido
La livedo reticularis no mejora a pesar del tratamiento anticoagulante, y no se ha descrito ningún tratamiento satisfactorio. Las necrosis cutáneas extensas y gangrenas digitales precisan tratamiento con AO a INR 2-3, como los pacientes con trombosis en otras localizaciones. Las lesiones pseudovasculíticas o de livedo vasculitis pueden responder a dosis bajas de AAS (100 mg/día) o antiagregantes plaquetarios. Si son intensas o recurrentes se podría considerar AO 16.
Actitud terapéutica durante el embarazo
No existen datos publicados que avalen la necesidad de tratamiento en mujeres con AcAF sin manifestación clínica alguna de SAF. De hecho, y dado que las pérdidas fetales tempranas (antes de la décima semana) de embarazos en mujeres sanas son muy frecuentes, no se aconseja la determinación de AcAF hasta que no hayan ocurrido al menos tres pérdidas tempranas inexplicadas y/o una pérdida tardía 12. Algunos autores recomiendan AAS a dosis bajas (75-100 mg/día) si hay aCL positivo o aCA IgG o B2-GP1 a títulos altos 17. Cuando la paciente tiene AcAF y antecedentes de morbilidad gestacional (dos o más pérdidas fetales tempranas, y/o una o más muertes fetales tardías), y/o ha sufrido algún fenómeno trombótico, se aconseja iniciar tratamiento con AAS a dosis bajas desde que se toma la decisión de concebir, y asociar heparina de bajo peso molecular (HBPM) en cuanto se confirma la gestación, manteniendo el tratamiento al menos hasta 6 semanas postparto 12. Y, por supuesto, requerirán un seguimiento y monitorización muy estrechos durante todo el embarazo para detectar una posible insuficiencia placentaria y retardo del crecimiento fetal 12,16,20. La AO puede provocar embriopatías, sobre todo en las semanas 6 a 12 de gestación. Las pacientes que ya están anticoaguladas por trombosis previas deben ser pasadas a HBPM tan pronto como se confirme la gestación y hasta el final de la misma 23.
Conflicto de intereses. Declaramos no tener ningún conflicto de intereses
Correspondencia:
Carmen García García.
Servicio de Dermatología. Hospital Universitario de La Princesa.
Diego de León, 62. 28006 Madrid.



