


INTRODUCCION
Las mucinosis papulares, clásicamente denominadas liquen mixedematoso, son un grupo de enfermedades caracterizado por el depósito de mucina en la dermis y la aparición de un grado variable de esclerosis en la piel en ausencia de disfunción tiroidea. La mucinosis papulosa acral persistente (MPAP) es un subtipo infrecuente de mucinosis papular en la que, por definición, no existe patología sistémica asociada. Presentamos el caso de una mujer sana de 52 años con lesiones cutáneas de MPAP en el dorso de ambas manos, cuyo estudio histológico reveló el depósito dérmico de mucina.
CASO CLINICO
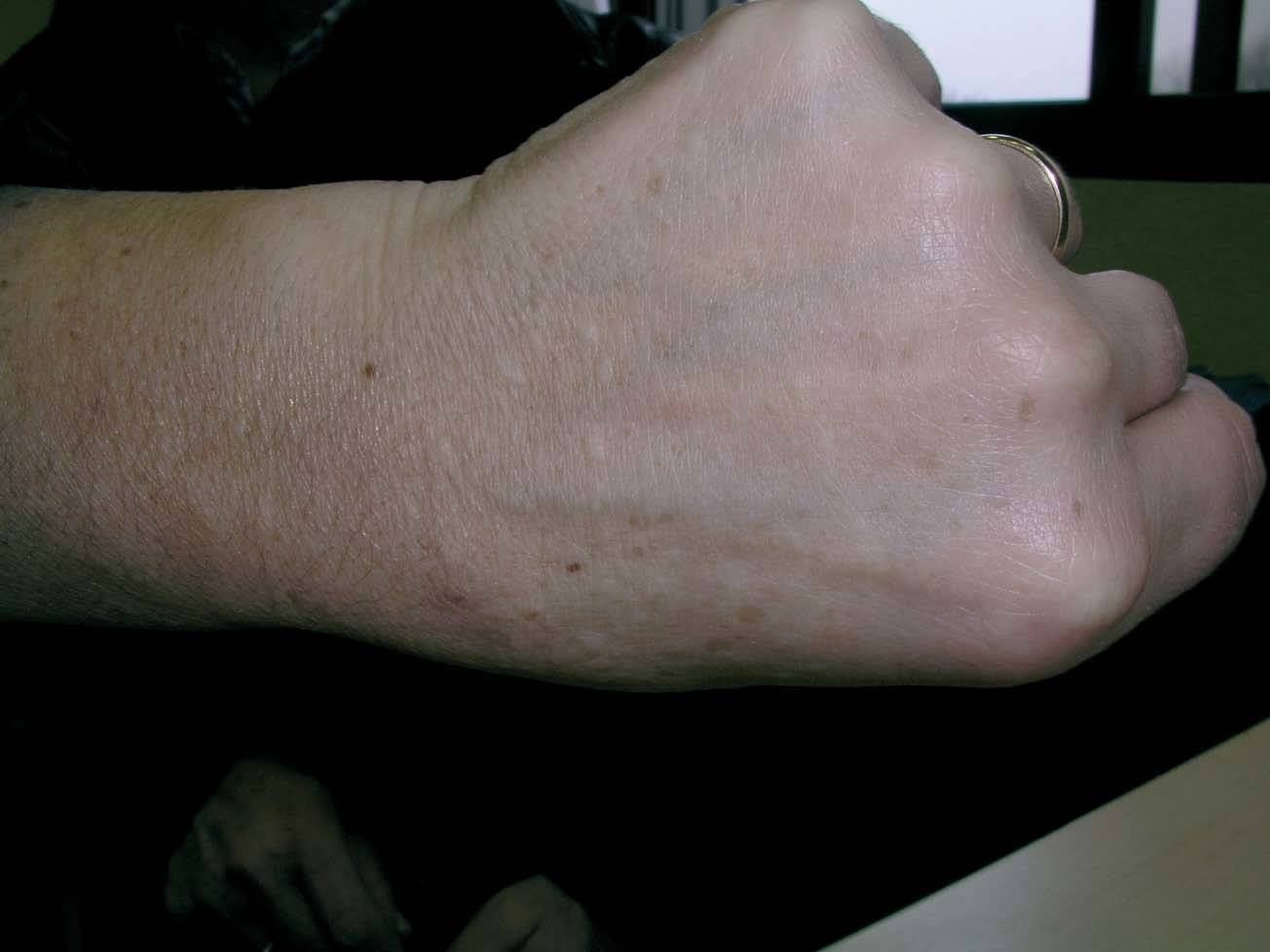
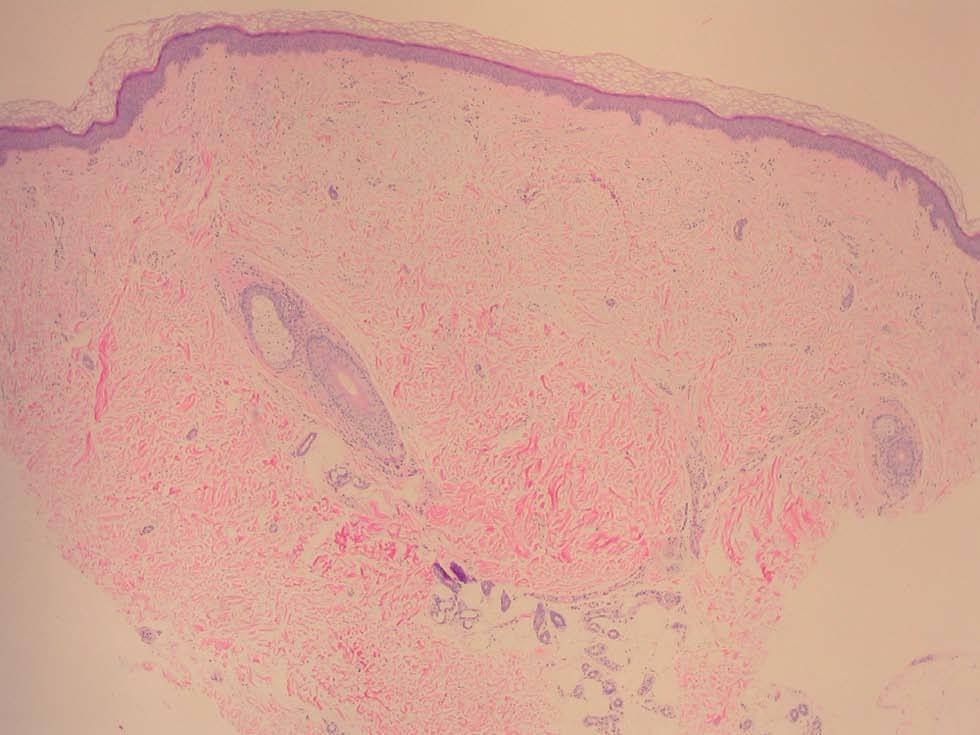
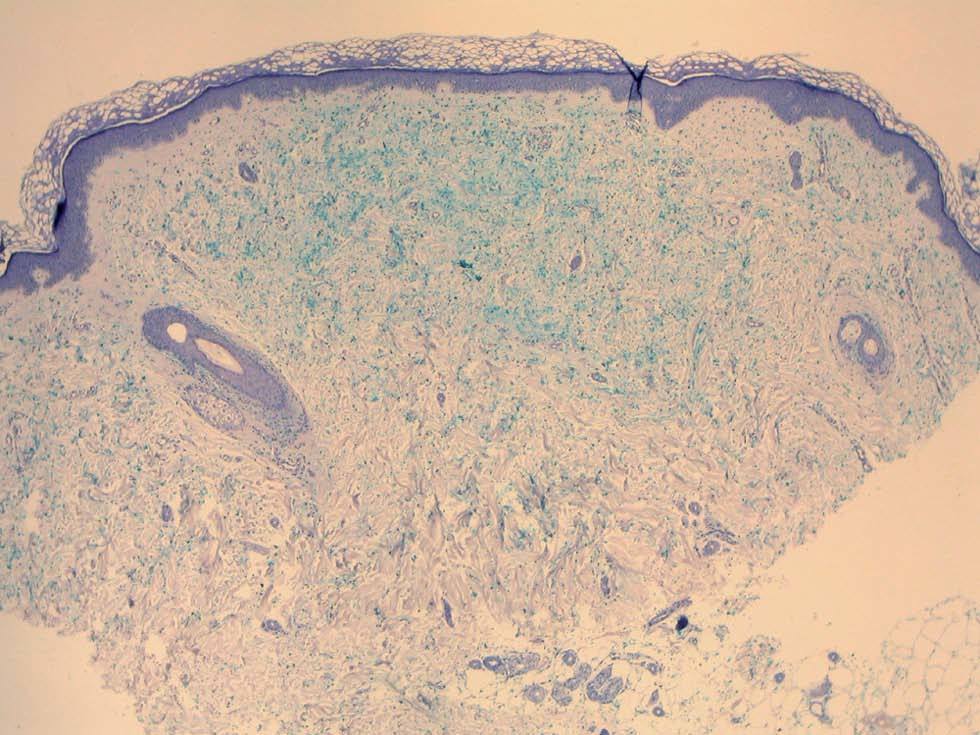
Una mujer de 52 años, sin antecedentes patológicos de interés, acudió a la consulta de dermatología de nuestro hospital por presentar lesiones cutáneas asintomáticas en el dorso de las manos desde hacía 4 años. Según la paciente, el proceso se había iniciado en el dorso de la mano derecha y había progresado lentamente hasta estabilizarse unos dos años después. En la exploración física se apreciaban lesiones papulosas firmes de tono blanquecino en el dorso de ambas manos y, de manera muy aislada, en la zona dorsal de las muñecas (fig. 1). Se realizó una biopsia cutánea de una de las lesiones, observándose una epidermis ligeramente atrófica y una llamativa separación de los tractos conectivos dérmicos por depósito de un material azul alcián positivo que correspondía a mucina (figs. 2 y 3). Se reconocía, además, una ligera rotura de fibras elásticas con técnica de orceína. El resultado de la biopsia fue histológicamente compatible con mucinosis cutánea. Todas las exploraciones analíticas fueron normales, incluyendo hemograma, pruebas de función hepática, renal y tiroidea, inmunoglobulinas en sangre y proteinograma. Hasta el momento, las lesiones permanecen estables.
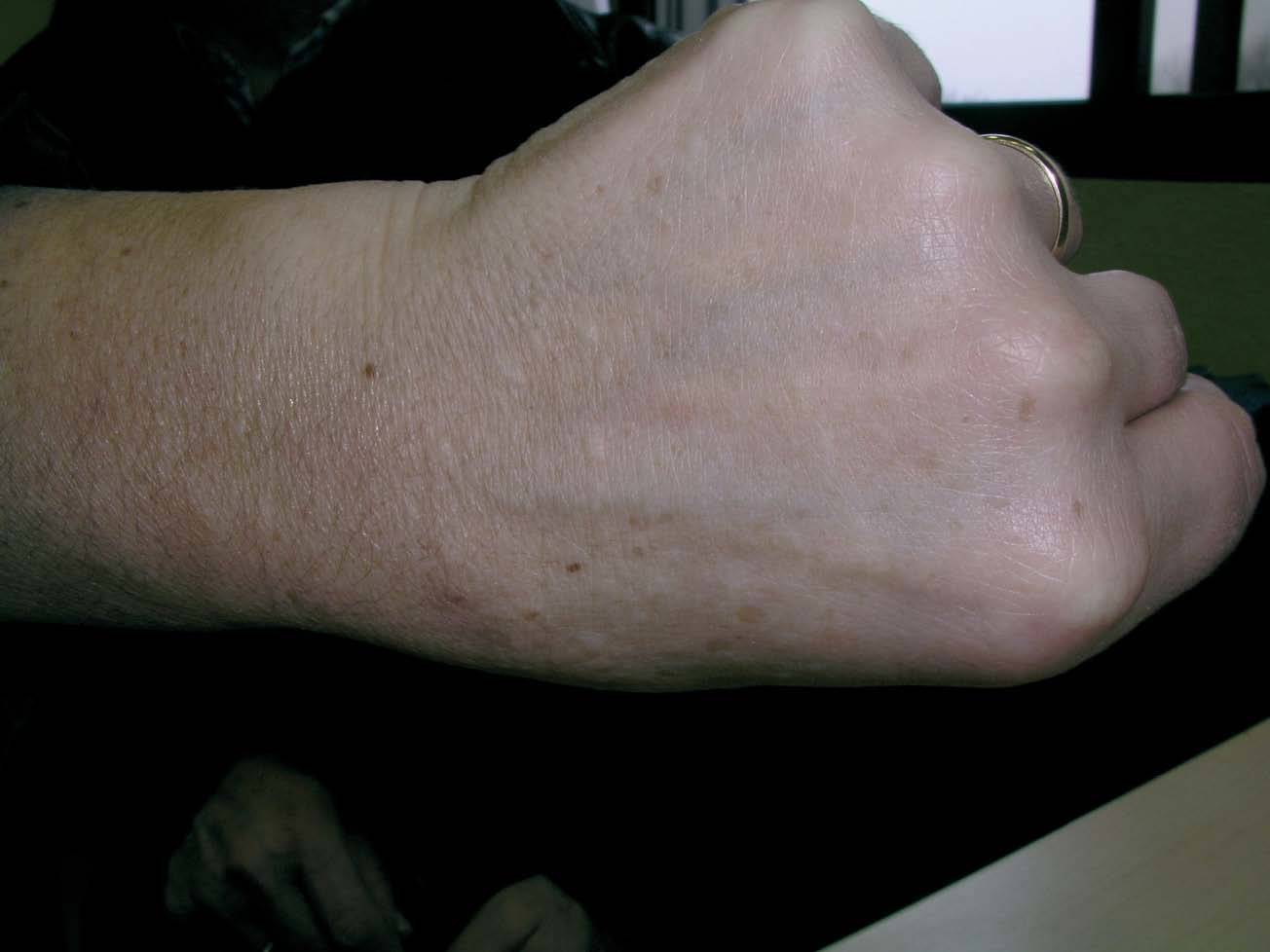
Fig.1.--Lesiones papulosas de tono blanquecino en el dorso de la mano izquierda.
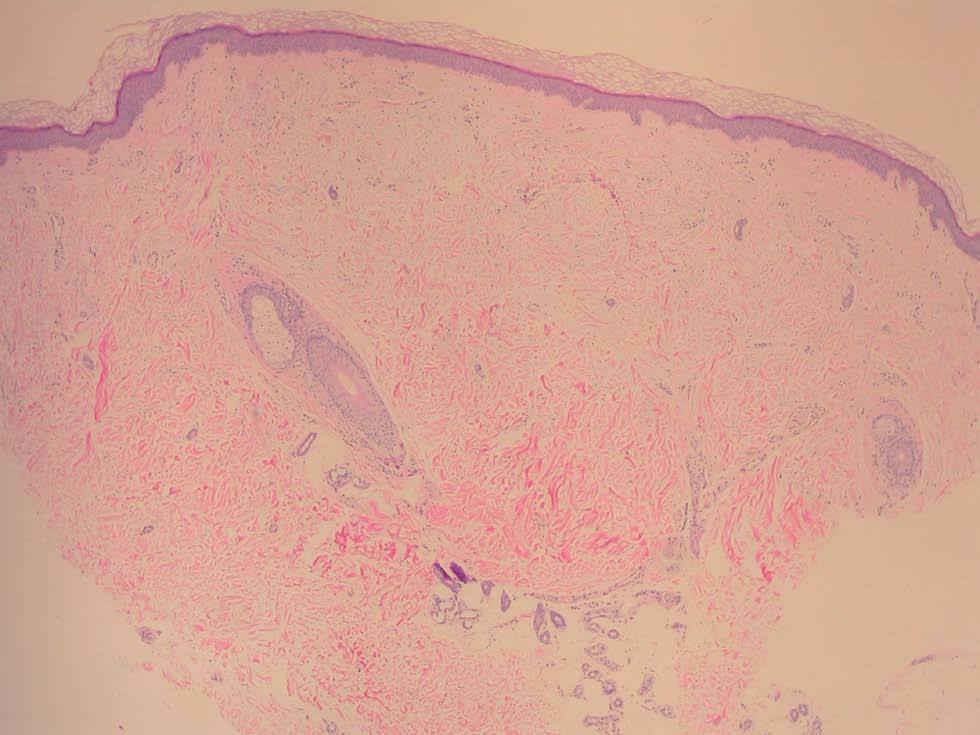
Fig. 2.--Pápula con ligera separación de las fibras colágenas dérmicas. (Hematoxilina-eosina, x 4.)
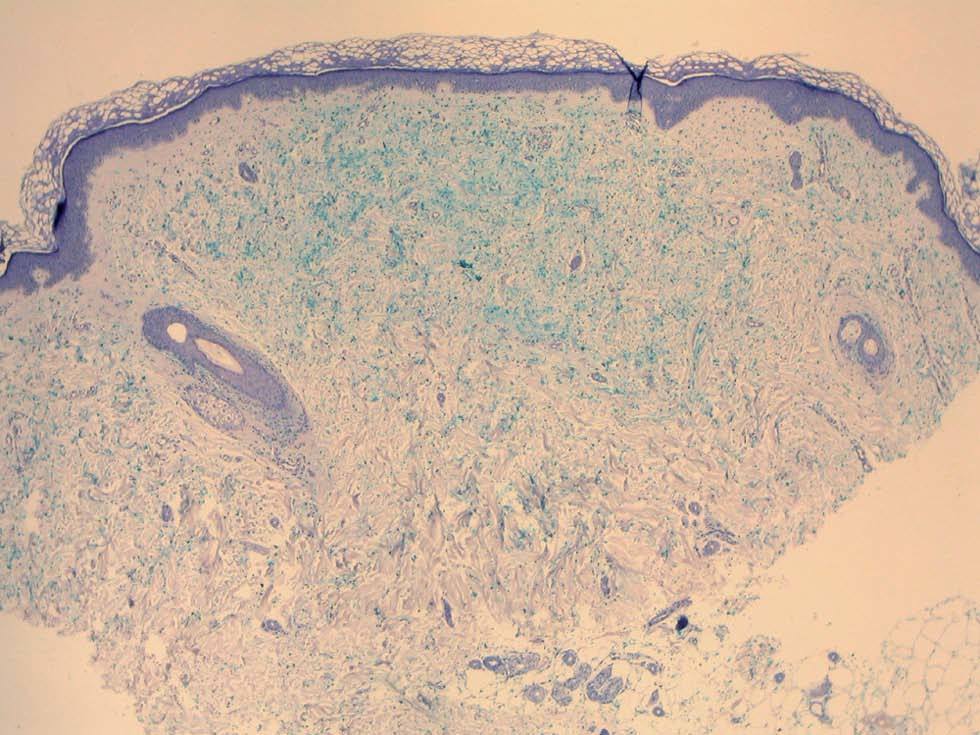
Fig. 3.--Depósito de mucina positivo con tinción de azul alcián. Una fina zona de Grenz separa el depósito de mucina de la epidermis. (Azul alcián, x 4.)
COMENTARIO
La clasificación más reciente de las mucinosis cutáneas fue publicada en el año 2001 por Rongioletti y Debora 1. Basándose en su propia experiencia y en la revisión de los casos publicados, estos autores dividen las mucinosis cutáneas en tres grupos: a) formas localizadas sin afectación sistémica (mucinosis papulares o liquen mixedematoso); b) formas generalizadas con afectación sistémica, fundamentalmente asociadas a gammapatías monoclonales tipo IgG (escleromixedema) y c) formas intermedias o atípicas que comparten criterios de los dos grupos anteriores, y que agrupan aquellos casos en los que las manifestaciones clínicas, la afectación sistémica, y/o el curso evolutivo de la enfermedad impiden la clasificación dentro de una entidad concreta. Dentro de las formas localizadas o mucinosis papulares existen cinco variantes clínico-patológicas, que incluyen la forma leve o discreta de mucinosis papulosa, la MPAP, la mucinosis papulosa autorresolutiva, la mucinosis papulosa de la infancia y la forma nodular 1. Nuestra paciente presentaba una forma localizada de mucinosis con afectación exclusiva del dorso de las manos y del tercio distal de los antebrazos, manifestaciones clínicas propias de la MPAP.
La mucinosis papulosa acral persistente fue descrita por primera vez en 1986 por Rongioletti et al 2. Dichos autores consideraban esta entidad como una forma diferenciada de mucinosis papular localizada, tanto desde el punto de vista clínico como desde el punto de vista histológico 3. Es más frecuente en mujeres, y clínicamente se caracteriza por la aparición de pápulas simétricas de 2 a 5 mm de diámetro, asintomáticas, que se localizan preferentemente en la zona extensora de las muñecas y en las manos. Histológicamente, se observa un depósito dérmico de mucina junto con una proliferación variable de fibroblastos. Las lesiones pueden aumentar en número con los años y, de acuerdo con los criterios diagnósticos que se detallan en la tabla 1, no debe existir una enfermedad sistémica asociada. No existe un tratamiento eficaz, aunque se ha visto una regresión espontánea de las pápulas en algunos casos. Recientemente se ha publicado un trabajo en el que se revisan todos los casos publicados hasta el momento de mucinosis acral papulosa persistente 4. Según este estudio, sólo hay 20 casos publicados que cumplan los criterios diagnósticos de MPAP. Sin embargo, dos de los pacientes incluidos en esta serie como «verdaderas MPAP» presentan anomalías sistémicas asociadas, incluyendo una gammapatía monoclonal tipo IgA kappa 5 y la presencia de anticuerpos antinucleares positivos 6, hallazgos que son interpretados en el estudio como asociaciones casuales sin relevancia etiopatogénica.

En resumen, presentamos un nuevo caso de MPAP, un raro tipo de mucinosis localizada de ubicación exclusivamente acral.
Declaración de conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Ángela Hernández.
Unidad de Dermatología. Hospital General Yagüe.
Avda. Cid, 96. 09005 Burgos. España.
ahernandez_hnj@yahoo.es
Recibido el 20 de febrero de 2006.
Aceptado el 26 de junio de 2006.



