




INTRODUCCION
Los pénfigos representan un grupo de enfermedades crónicas de la piel y las mucosas, de fisiopatogenia autoinmune, que se caracterizan clínicamente por la presencia de ampollas intraepidérmicas cuyo mecanismo de formación es una pérdida de cohesión entre los queratinocitos epidérmicos conocido como acantolisis. Inmunopatológicamente estos pacientes presentan autoanticuerpos intercelulares circulantes (AcIC) de tipo inmunoglobulina (Ig) G en el suero, dirigidos contra un epítope localizado en los desmosomas de los queratinocitos de los epitelios planos estratificados. Las formas más frecuentes de este grupo son el pénfigo vulgar, el pénfigo foliáceo y el pénfigo paraneoplásico.
El pénfigo vulgar (PV) es la forma más frecuente y severa de pénfigo en los países desarrollados, representando el 80 % de todos los casos 1. Se trata de una enfermedad cuya característica principal es el depósito de autoanticuerpos de tipo IgG y ocasionalmente de tipo IgA, contra la desmogleína 3 (Dgs 3), la cual es una glucoproteína desmosomal perteneciente a la familia de las cadherinas 2. Estas son moléculas de adhesión celular que forman parte de los desmosomas y que proporcionan las conexiones físicas entre los queratinocitos 3,4. Los desmosomas epidérmicos contienen fundamentalmente Dgs 1 y Dgs 3, esta última es el antígeno reconocido por todos los sueros de pacientes con PV. Estos autoanticuerpos frente a la desmogleína 3 están implicados en la patogenia de esta enfermedad y sus niveles se correlacionan con el grado de actividad de la misma 5.
Dentro de los factores genéticos que influyen en la patogénesis del PV, uno de los más relevantes es el complejo mayor de histocompatibilidad (CMH), que es una región de genes polimórficos, localizados en el brazo corto del cromosoma 6, cuyos productos se expresan en la superficie de una gran variedad de células.
Se ha descrito una predisposición genética a padecer pénfigo 6,7 ligada al sistema de antígenos de histocompatibilidad (HLA), destacándose dos haplotipos: DRB1*0402 (HLA DR4) DQB1*0302 (HLA DQ8) y DRB1*1401 (HLA DR14) DQB1*0503 (HLA DQ5) 8-10.
A pesar de que puede observarse en todos los grupos étnicos y raciales, existe una mayor incidencia de PV en los judíos 11,12. En los judíos Ashkenazi los tipos del haplotipo HLA DRB1*0402 y DRB1*1402 son mucho más frecuentes que en otras razas (92,3 %) 8,13-17.
Basándonos en que existe una susceptibilidad genética a padecer pénfigo ligada al HLA, decidimos realizar un estudio en tres familias zamoranas diagnosticadas de PV familiar, con el objetivo de determinar la frecuencia de los antígenos HLA de clase II junto con el riesgo relativo (RR) que confieren dichos antígenos a padecer la enfermedad y posteriormente describir las variantes alélicas de clase II DR y DQ más frecuentes en dichas familias.
MATERIAL Y MÉTODO
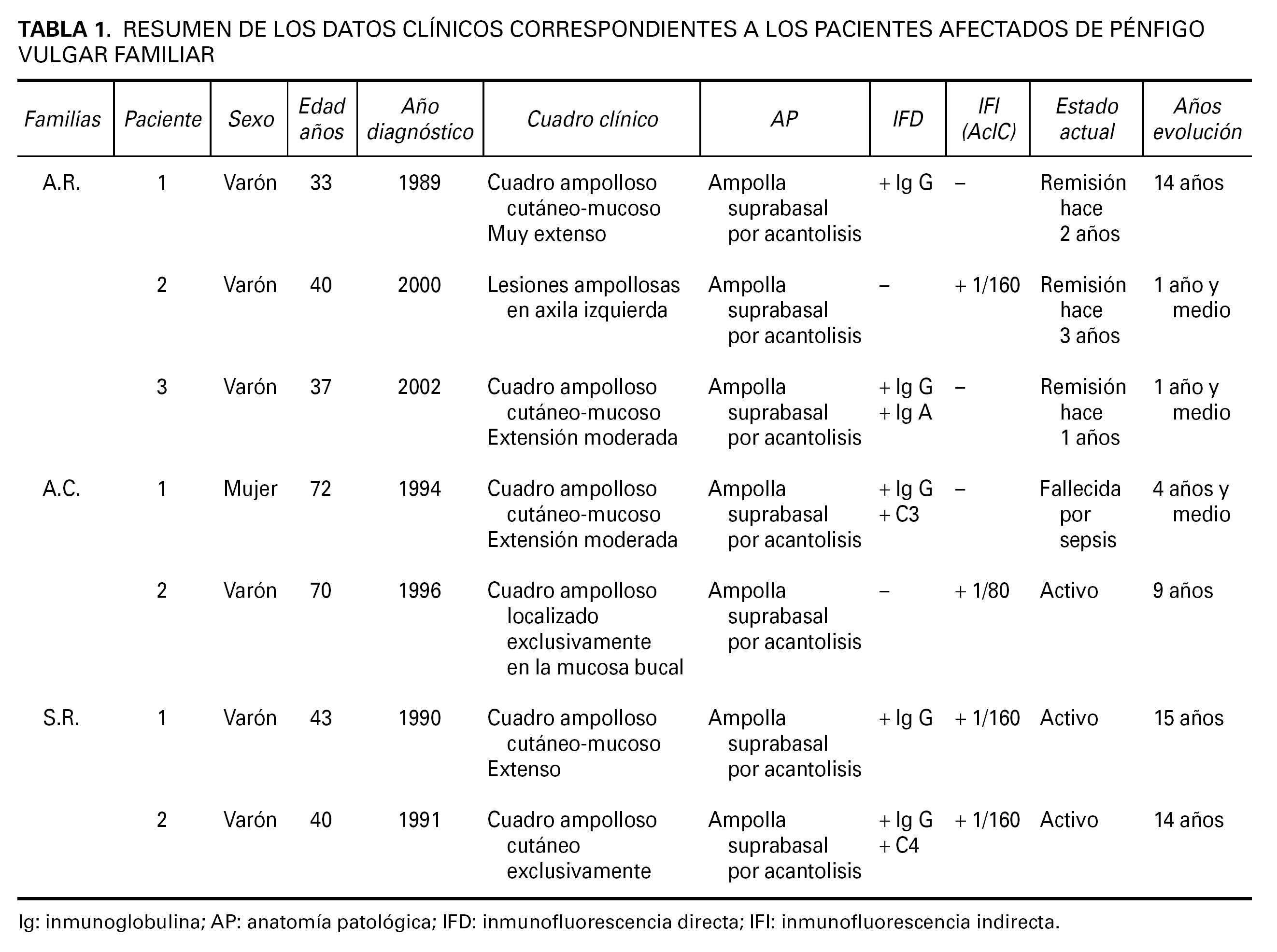
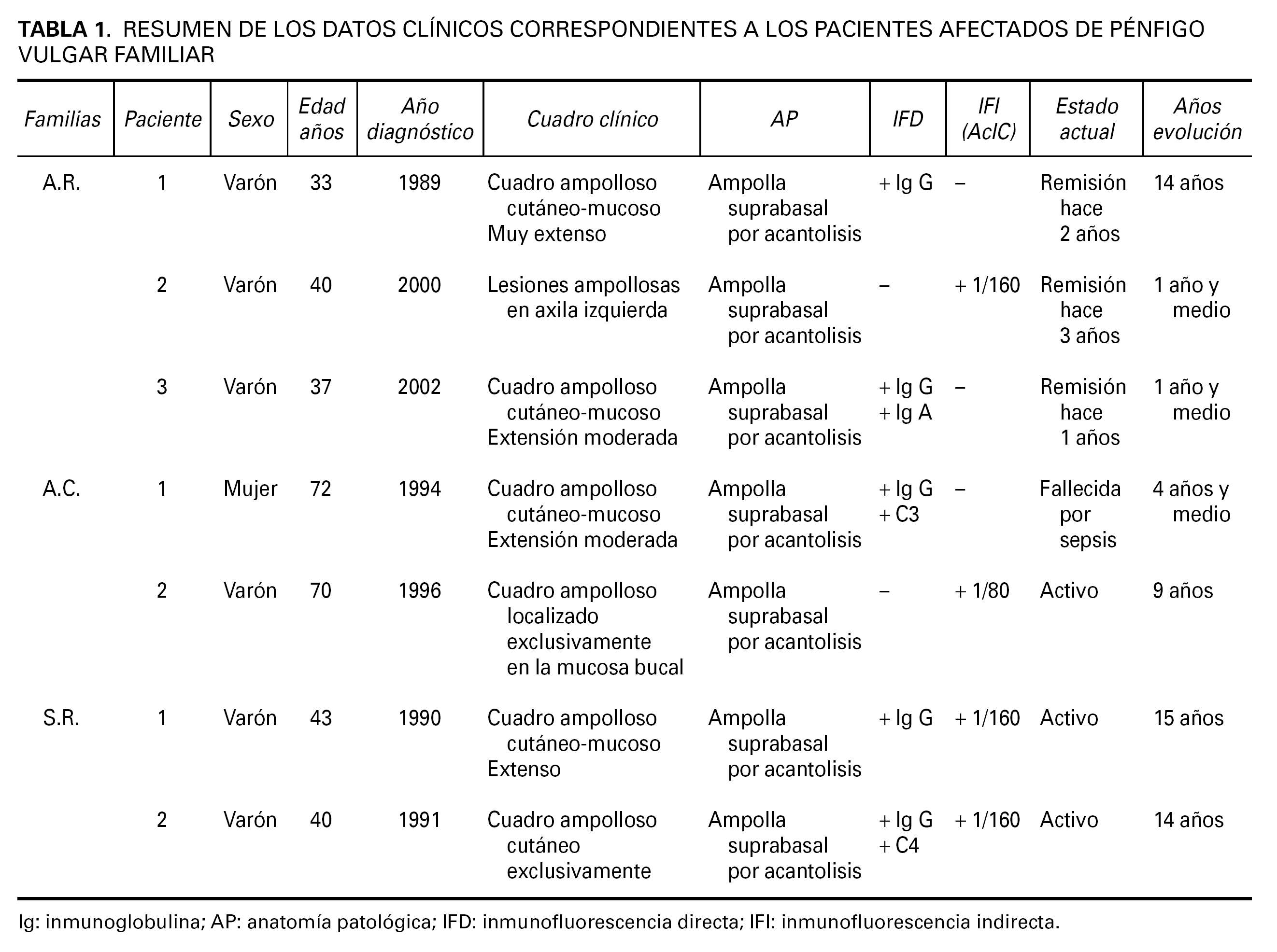
Se revisaron de forma retrospectiva tres familias, no emparentadas entre sí, con historia de varios miembros afectados de PV atendidas en el Servicio de Dermatología de Zamora entre el período 1989-2002. Encontramos 7 pacientes que cumplían criterios diagnósticos de PV familiar tanto clínicos (presencia de ampollas a nivel cutáneo y/o mucoso) como histopatológicos (ampollas supra-basales con acantolisis). Diagnóstico que se corroboró mediante estudios de inmunofluorescencia directa y/o indirecta, poniendo de manifiesto la presencia de depósitos de IgG en los espacios intercelulares de la epidermis afecta y de AcIC en suero respectivamente.
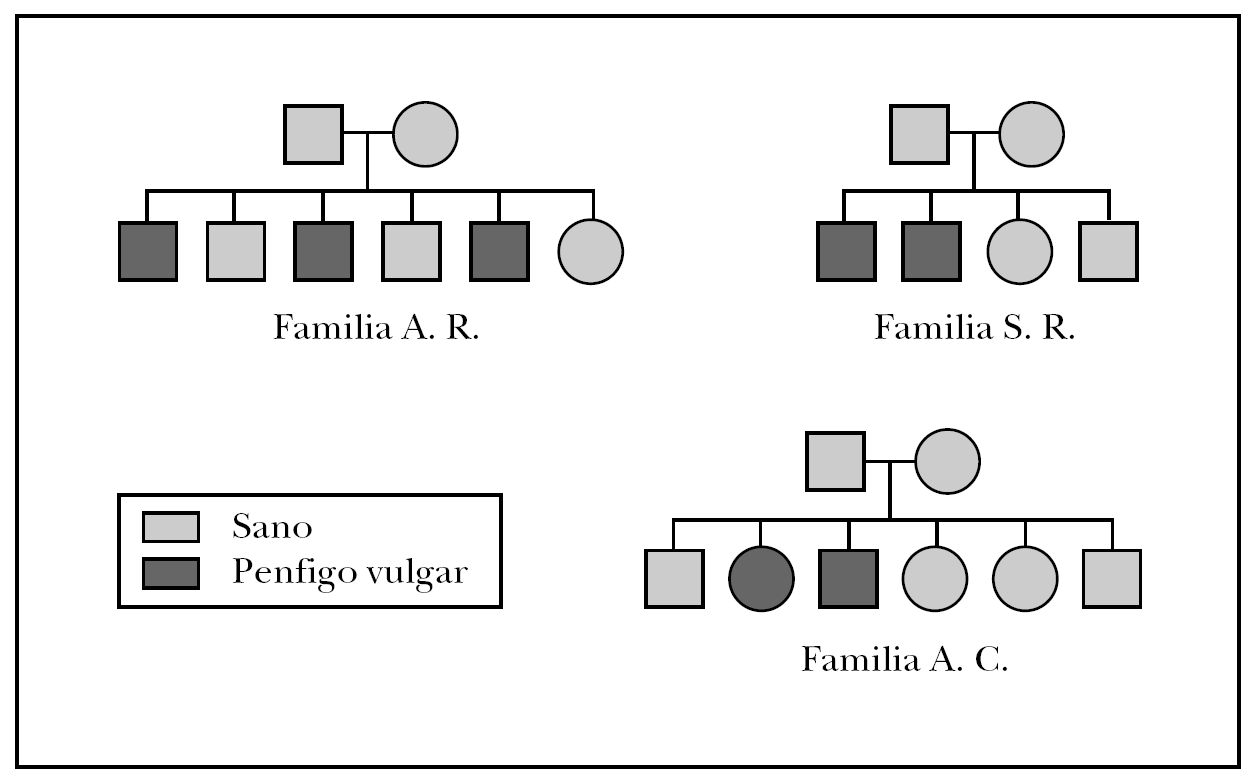
Las familias que formaron parte del estudio fueron: la familia A.R., formada por 6 hermanos, de los cuales tres de ellos padecían PV, la familia S.R., con 4 familiares directos dos de ellos diagnosticados de PV y la famila A.C., compuesta por 6 familiares directos de los cuales dos de ellos habían sido diagnosticados de PV. El árbol genealógico de las tres familias del estudio se muestra en la figura 1.
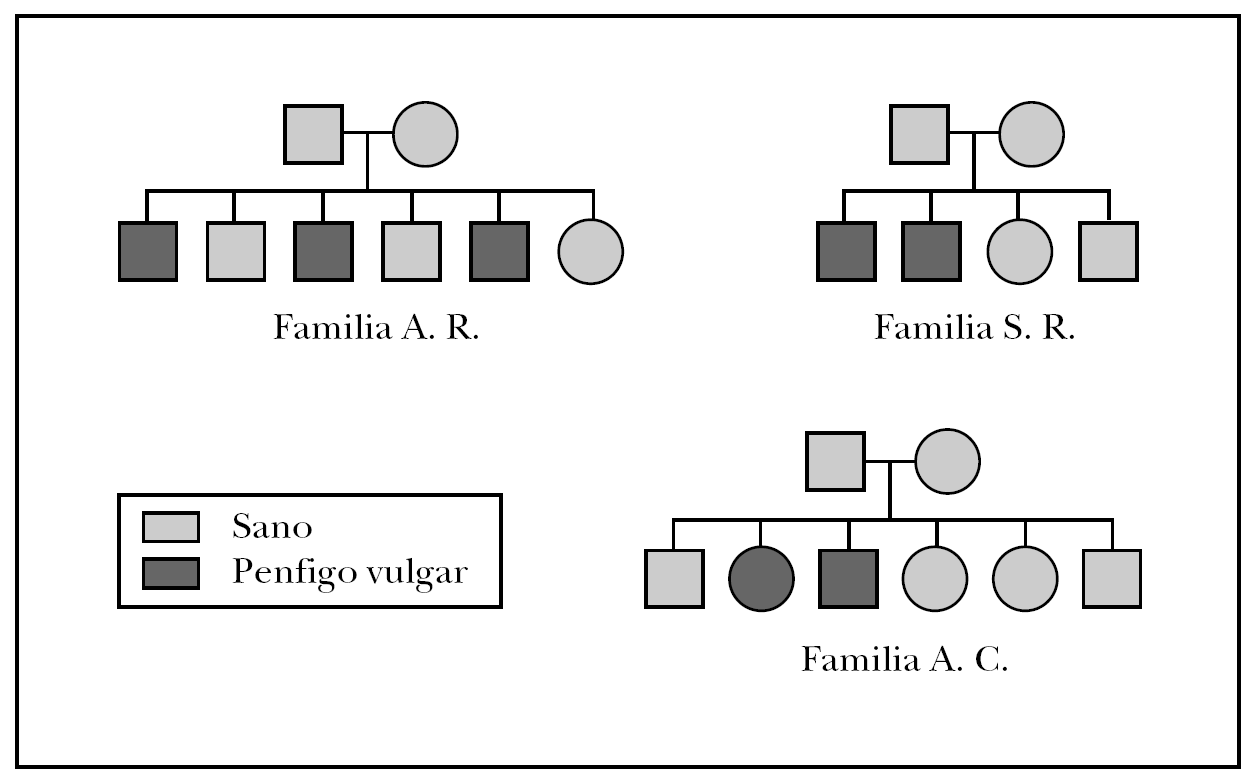
Fig. 1.--Árbol genealógico de las familias afectadas de pénfigo vulgar familiar.
Los datos clínicos de los pacientes afectados de PV familiar referentes al cuadro clínico, año de diagnóstico y evolución se resumen en la tabla 1.
Se realizó un estudio observacional y transversal de las tres familias diagnosticadas de PV familiar, con el objetivo de determinar los genes presentes en los loci DQB1 y DRB1 de todos los sujetos vivos de ambas familias, tanto de los pacientes con PV como de los sanos. Para ello se tomó una muestra de sangre periférica para extracción del ADN genómico de los leucocitos mediante la técnica de fenol. Posteriormente se realizó una determinación genética de los alelos de los locus de HLA-DR y HLA-DQ amplificando las regiones HLA-DRB1 y HLA-DQB1, mediante la técnica de la reacción en cadena de la polimerasa (PCR) con tipificación mediante iniciadores o primers específicos (PCR-SSP, kit de alta resolución para DQB1 y de baja resolución para DRB1).
RESULTADOS
Las 3 familias diagnosticadas de PV familiar estaban compuestas de 16 miembros, de los cuales 7 de ellos habían sido diagnosticados de PV, y los 9 restantes no habían presentado en ningún momento clínica compatible con dicha enfermedad ampollosa. Todos ellos carecían de historia familiar y/o personal de otras enfermedades autoinmunes.
Se incluyeron en el estudio 9 personas, de las cuales 6 padecían PV familiar, uno de los pacientes afectados no pudo introducirse en el estudio por fallecimiento como consecuencia de una sepsis. Las 7 personas restantes no pudieron ser estudiadas por no residir en nuestra provincia o por fallecimiento.
Posteriormente realizamos el estudio genético a todas las personas incluidas en el estudio. Los genes presentes en los locus DQB1 y DRB1 de todos ellos se muestran en las tablas 2-4.



Todos nuestros enfermos de PV tenían alelos que codifican antígenos HLA DR 4 y DR 14.
DISCUSION
El PV familiar es excepcional, y solamente se ha descrito en la literatura médica afectación de familiares de primer grado 18,19. La observación de estos casos familiares, en los que se ven afectados varios miembros de una misma familia, demuestra una vez más la importancia que los factores genéticos tienen en la patogenia de dicha enfermedad.
En una exhaustiva revisión bibliográfica realizada hemos encontrado 34 familias con 71 miembros afectados. En 1991 Feinstein et al 20 publicaron una revisión con 25 familias y 53 miembros afectados de PV.
La asociación con un alelo de HLA específico implica que el estímulo inmunogenético para la autoinmunidad sería un péptido específico, y que al menos esta autoinmunidad dependería inicialmente de la activación de uno o varios clones de células T potencialmente autoagresivas. La asociación entre genes de HLA clase II y el PV ha sido muy bien documentada y las técnicas de análisis moleculares de la región HLA DP, DQ y DR han revelado que esta asociación se correlaciona con alelos muy específicos. En concreto, los alelos DRB1*0402 y/o DQB1*0503 son los más frecuentemente asociados en los diversos grupos étnicos 9,16,21.
Los antígenos que se han propuesto como más importantes en esta predisposición genética son DR4 y/o DR14. El gen DR4 es el que comúnmente se ha asociado en la población judía (está presente en el 90 % de los judíos y especialmente en los Ashkenazi) 13,22. En los pacientes caucasianos no judíos se ha visto un incremento de la frecuencia de HLA DR4, DR14, DQ8, DR6 Y DQ5 23,24.
El inicio y desarrollo del PV puede resultar de la interacción de factores externos y factores genéticos, especialmente alelos del CMH. Como hoy sabemos, estos alelos desempeñan un papel importante en la susceptibilidad para padecer la enfermedad. Hay estudios que indican que determinadas posiciones de aminoácidos en las moléculas DR4 y DR14 aumentan su capacidad para fijar selectivamente porciones de desmogleína, originando linfocitos autorreactivos. Por todo ello es trascendente identificar aquellos que se asocian con mayor frecuencia y que confieren una mayor predisposición a padecer PV con el fin de diseñar en un futuro estrategias inmunoterapéuticas.
En el presente estudio se demuestra una asociación significativa entre el PV familiar y los antígenos de clase II del CMH, especialmente con DR 4 y DR 14. En cuanto a los genes que codifican estos antígenos los más positivamente asociados con PV forman parte de dos haplotipos diferentes: DRB1*04 DQB1*0302/11 y DRB1*14 DQB1*0503, que confieren un mayor RR para padecer dicha enfermedad (RR 14,4 %).
Los tres hermanos de la familia A.R. afectados de PV presentan idénticos haplotipos (DRB1*04; DQB1* 0302/11 y DRB1*14; DQB1*0503). Este haplotipo también es compartido por uno de los hermanos que no padece PV, teniendo una posibilidad muy alta de desarrollarlo, aunque no ha presentado hasta la actualidad clínica compatible con dicha enfermedad y la determinación de los autoanticuerpos ha sido negativa. Otro de los hermanos, que no presenta la enfermedad, comparte el DRB1*14 y DQB1*0503 con los hermanos afectados, de manera que la probabilidad de desarrollarla es alta, y por último la hermana no afectada de pénfigo comparte HLA DRB1*04; DQB1* 0302/11 con los afectados por ello también su probabilidad de desarrollarla es alta.
En la familia S.R, los dos hermanos varones afectados de PV tenían un haplotipo HLA DRB1*04; DQB1* 0302/11 y HLA DRB1*14; DQB1*0302/11. Son homocigotos para el locus DQB1 y tienen la mitad del haplotipo igual a los pacientes afectos de la familia A.R.
El único enfermo estudiado de la familia A.C tenía un haplotipo HLA DRB1*04; DQB1*0302/11 y HLA DRB1*14; DQB1*0503, exactamente igual a los pacientes con PV de la familia A.R.
Después de este análisis molecular queda claro que el RR conferido por la presencia de HLA DR4 y/o DR14 en el desarrollo de PV es muy elevado, como se demuestra en nuestros pacientes. La mayoría de los pacientes descritos en la literatura tienen alguno de estos dos alelos, en la población italiana y japonesa aproximadamente la relación es de 2:3 para los que tienen HLA DR14 y 1:3 para HLA DR4, proporción que se invierte en los judíos Ashkenazi y en los españoles 19.
El porcentaje de pacientes que presentan los dos alelos HLA DR14 y DR4 es inferior, oscilando entre un 11,5 % 15 y un 22,6 % 1, contrastando con el elevado porcentaje observado en nuestra serie (100 %). La presencia de ambos alelos en algunas familias como las aquí estudiadas, justificaría el aumento de la predisposición a padecer PV, pudiéndose observar varios miembros con dicha enfermedad, como sucede en la familia A.R que presenta una elevada incidencia de la enfermedad (el 50 % de sus miembros están afectados).
Los 6 pacientes con PV que hemos estudiado presentaban el alelo DQB1* 0302 y 4 de ellos el alelo DQB1* 0503, esto se sabe que es por un desequilibrio de unión con DRB1* 04 y 14 respectivamente, es decir, se heredan juntos no siendo significativos por sí mismos.
No todos los pacientes portadores de HLA DR4 o DR14 padecen PV, aunque el RR de padecer la enfermedad teniendo alguna de estas dos moléculas sea elevado (14,4 %). Esto nos hace pensar que los factores inmunogenéticos, a pesar de tener una importancia básica en la patogenia de la enfermedad, no se consideran suficientes por sí mismos para su desarrollo. Se necesita además una serie de factores adicionales que actúen sobre el complejo mecanismo de regulación de la expresión de las moléculas HLA de clase II en la superficie de las células, para que los individuos con una determinada predisposición genética a desarrollar pénfigo manifiesten clínicamente la enfermedad 25. Algunos de estos factores son conocidos clásicamente como ciertos medicamentos, alimentos ricos en grupos thiol, exposición solar, quemaduras, etc., pero otros muchos están aún por descubrir.
Contrastando nuestros datos con el estudio de González-Escribano et al 10, podemos concluir que, en la población española afectada de PV el HLA DR4 y el HLA DR14 están involucrados en la patogénesis de esta enfermedad. En nuestro estudio, nos llamó la atención que pese a ser un número pequeño de pacientes, se observó una fuerte asociación con los alelos DQB1*0302 y DQB1*0503, presentes en el 83,3 % y en el 66,67 % de los pacientes respectivamente. Con relación al alelo DQB1*0302 el porcentaje es superponible al estudio español de González-Escribano et al que es de un 88,5 %, pero sin embargo el porcentaje asociado a DQB1*0503 es ligeramente inferior al de nuestro estudio, un 30,8 %. El genotipo DRB1*0302 DRB1*0503 fue hallado en varios pacientes con PV, lo que sugiere un sinergismo entre ambos haplotipos para conferir susceptibilidad. Estas variantes alélicas también se han asociado en los judíos Askenazi 13, japoneses 8, indios 16 e italianos 9. Sin embargo, creemos necesario el estudio de otros factores, principalmente de tipo ambiental, para comprender la forma en que interactúan para matizar la expresión clínica de la enfermedad en pacientes genéticamente predispuestos.
Declaración de conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
M.ª Teresa Bordel Gómez. Servicio de Dermatología.
Complejo Asistencial Virgen de la Concha. Martínez Villergas, 6, 1.ºB. 49003. Zamora. España.
matebordel@yahoo.es, maitebordel@aedv.es
Recibido el 27 de enero de 2006.
Aceptado el 17 de Julio de 2006.



