

INTRODUCCION
El sarcoma epitelioide, descrito por Enzinger en 19701, está considerado, pese a su rareza, como el tumor maligno de tejidos blandos más frecuente de la mano y la muñeca. Afecta preferentemente a varones jóvenes y se presenta como un nódulo solitario o múltiple de 3 a 6 cm de diámetro, en general asintomático y de crecimiento lento que puede adherirse al tendón y con frecuencia se ulcera. Su aspecto clínico es inespecífico, lo que justifica el retraso diagnóstico. La histopatología y características inmunohistoquímicas permiten su identificación en las formas evolucionadas, pero en sus estadios iniciales y en algunas variantes es necesario establecer el diagnóstico diferencial con múltiples procesos. El tratamiento de elección es la cirugía agresiva, que puede complementarse con radioterapia y/o quimioterapia. A pesar de ello, el porcentaje de recurrencias es alto y tiene una elevada capacidad metastásica.
Se presenta un caso que pone de manifiesto las dificultades diagnósticas que, en ocasiones, puede plantear así como su agresividad.
DESCRIPCION DEL CASO
Un varón de 27 años presentaba una tumoración subcutánea dolorosa en el antebrazo derecho de 6 meses de evolución. La lesión se había desarrollado sobre la cicatriz tras la extirpación, en otro centro, 2 años antes, de una lesión que había sido diagnosticada de fascitis nodular seudosarcomatosa.
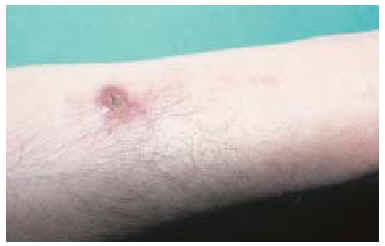
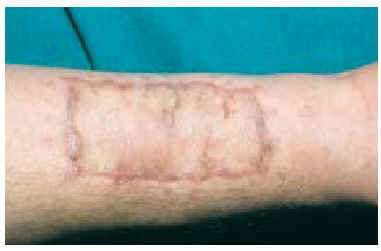
En la exploración clínica presentaba, en el tercio distal de la cara flexora del antebrazo derecho, una cicatriz longitudinal de 2,5 cm, en cuyo extremo existía una tumoración nodular eritematosa de 1,5 cm de diámetro, ulcerada, mal delimitada, de consistencia dura y superficie multilobulada (fig. 1). La lesión estaba adherida a planos profundos y era dolorosa a la palpación. No se palpaban adenopatías epitrocleares, axilares o supraclaviculares. La analítica general y el estudio radiológico de brazo y tórax fueron normales.
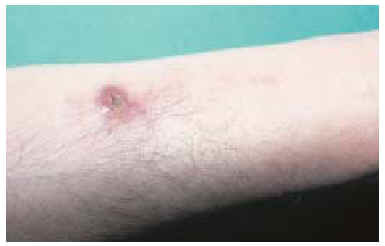
Fig. 1.--Tumoración nodular ulcerada en el extremo de la cicatriz de una extirpación previa.
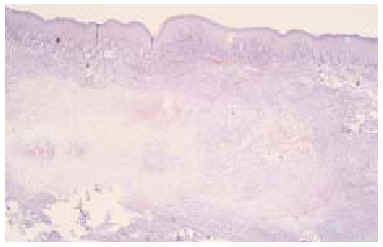
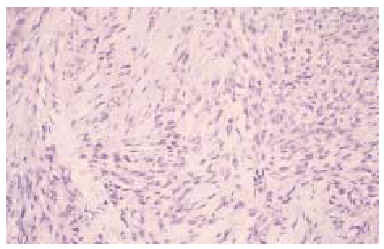
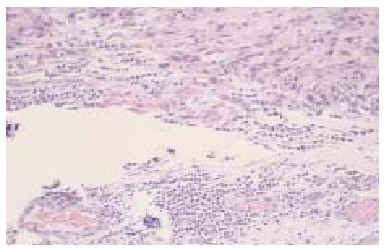
Se realizó una biopsia «en sacabocados» de la lesión, que mostró un proceso inflamatorio necrosante inespecífico, que se informó como un absceso fistulizado, por lo que se practicaron cultivos microbiológicos y micológicos que sólo demostraron patógenos contaminantes. Tras reconsiderar el caso, se realizó una biopsia incisional más amplia, observándose rasgos histopatológicos característicos de sarcoma epitelioide, que más tarde fueron confirmados tras el estudio de la pieza operatoria. En el estudio con hematoxilina-eosina se observaba una proliferación neoplásica mesenquimatosa que se disponía como agregados nodulares alrededor de áreas de necrosis y de degeneración del colágeno conformando un patrón de falsos granulomas «en empalizada». La lesión ocupaba toda la dermis y tejido celular subcutáneo, y en su zona central contactaba con la epidermis, ulcerándola (fig. 2). La celularidad del tumor presentaba zonas con predominio de células de aspecto fusiforme, pleomórficas, con numerosas mitosis, y otras zonas donde predominaban células de hábito epitelioide, con núcleo grande y heterocromatina muy dispersa y en grumos, con varios nucléolos y un citoplasma eosinófilo (fig. 3). Las células tumorales se disponían alrededor de áreas de necrosis y de necrobiosis a modo de empalizada periférica, y estaban rodeadas de un infiltrado inflamatorio denso de predominio linfocitario. En el plano profundo y yuxtafascial de la lesión, las células epitelioides se disponían en banda, en pequeños grupos o como células aisladas, rodeadas de un estroma fibroso (fig. 4). Se observaron también zonas tumorales con predominio de células de características rabdoides con gran citoplasma globular eosinófilo y núcleo excéntrico con gran atipia celular. El estudio inmunohistoquímico mostró positividad para citoqueratinas de alto y bajo peso molecular, antígeno de membrana embrionario (EMA) y vimentina. El resto de marcadores realizados (S-100, HMB-45, desmina, enolasa, neurofilamentos, actina y miosina) resultaron negativos.
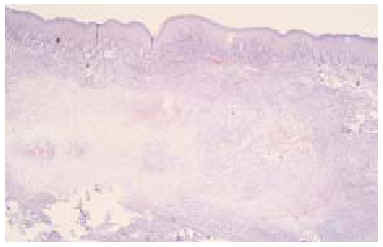
Fig. 2.--Proliferación celular con un patrón de seudogranulomas que ocupa toda la dermis e hipodermis y contacta en algunas zonas con la epidermis. (Hematoxilina-eosina, x40.)
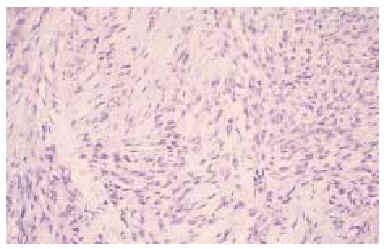
Fig. 3.--Células fusiformes pleomórficas con mitosis junto a otras de aspecto epitelioide. (Hematoxilina-eosina, x100.)
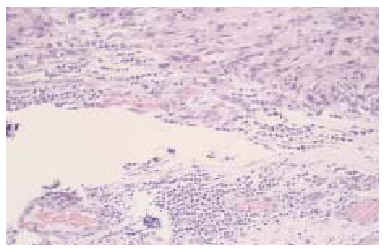
Fig. 4.--Disposición aislada o en pequeños grupos de las células tumorales, rodeadas de estroma fibrosa y presencia de cierto infiltrado inflamatorio linfocitario. (Hematoxilina-eosina, x100.)
Tras la confirmación diagnóstica, tuvimos la oportunidad de revisar la histopatología de la pieza operatoria de la primera intervención, realizada 2 años antes y en otro centro. Se observaba gran cantidad de células fusiformes con abundantes mitosis sobre un estroma fibroso con un mínimo componente de células epitelioides. No había necrosis ni patrón en empalizada. Probablemente, el aspecto fibroso reactivo y la baja agresividad de la lesión, hicieron que se diagnosticara de fascitis nodular seudosarcomatosa. Se concluyó que se trataba del mismo tumor, concretamente de la variante «similar a un fibroma» de sarcoma epitelioide, cuya característica principal es el aspecto fibromatoso de las lesiones primitivas, variando la celularidad en las recidivas hasta adquirir las características de sarcoma epitelioide clásico que presentaba en los estudios histopatológicos posteriores.
Tras información al paciente, se realizó extirpación, con 3 cm de margen superficial y fascia muscular en profundidad, de la lesión de la cicatriz anterior y cierre del defecto resultante mediante injerto de piel laminar. Histológicamente se comprobó que los márgenes tumorales, tanto profundos como superficiales, estaban libres de tumor. A los 6 meses de la intervención se detectó en la periferia de la zona injertada la presencia de varios nódulos subcutáneos, de consistencia dura (fig. 5). El estudio mediante punción-aspiración con aguja fina (PAAF) confirmó la presencia de células compatibles con recidiva local del tumor. Tras estudio de extensión negativo se decidió plantear al paciente la amputación del miembro como tratamiento más adecuado para intentar controlar la enfermedad primaria. El paciente rechazó la intervención, y no volvió a consultar, por lo que se desconoce su evolución y situación actual.
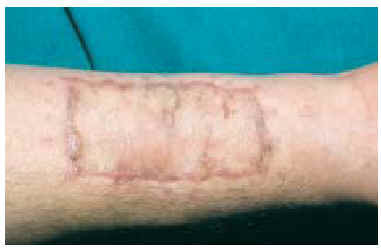
Fig. 5.--Aspecto a los 6 meses de la intervención. En la periferia del injerto se palpan nódulos que ponen de manifiesto la recidiva tumoral.
DISCUSION
El sarcoma epitelioide es una neoplasia de crecimiento lento que tiene su mayor prevalencia en adolescentes y adultos jóvenes entre los 10 y los 35 años, con una media de 26 años, doble incidencia en varones que en mujeres y sin diferencias interraciales1-4. Se localiza con más frecuencia en la zona distal de las extremidades superiores, en concreto en dedos, mano y antebrazo, aunque también se puede localizar en rodilla, región pretibial, nalgas y muslos. Otras localizaciones posibles son el cuero cabelludo, la vulva y el pene2,3. La posibilidad de localización sobre zonas sometidas a traumatismos repetidos o cicatrices antiguas pueden hacer pensar en estos sucesos como factores promotores en la génesis del sarcoma epitelioide5. La histogénesis del sarcoma epitelioide es desconocida. Se le supone un origen mesenquimatoso y una posterior metaplasia hacia células de estirpe epitelial6,7.
Clínicamente aparece como un nódulo o masa de consistencia dura, de 1-5 cm de diámetro. Su profundidad es variable. A veces crece en superficie y puede ulcerar la epidermis. En la mayoría de los casos infiltra dermis y/o hipodermis, y puede profundizar hasta adherirse a la fascia o al tendón, lo que origina una tumoración peor delimitada en forma de masa o placa multinodular que se desplaza con los movimientos articulares. El curso es indolente, aunque algunos pacientes refieren dolor a la palpación o a la movilización del miembro. El estudio radiológico es inespecífico, y muestra una masa de tejidos blandos que a veces contiene depósitos de calcio y que en algunos casos infiltra el hueso adyacente1. A pesar de tener un crecimiento lento, son tumores extremadamente agresivos, con un alto índice de recurrencias locales tras la extirpación y un gran poder metastatizante.
Para el diagnóstico de certeza es necesario el estudio histopatológico, aunque, en ocasiones se requieren varias biopsias en diferentes momentos evolutivos. Es muy característico el patrón nodular coalescente de las células tumorales que afectan a la dermis y al tejido celular subcutáneos, si bien también pueden afectar a la epidermis ulcerándola e infiltrar estructuras más profundas2,8. La celularidad está constituida por una proporción variable de células de morfología fusiforme y células epitelioides poligonales, con núcleo grande y citoplasma amplio, muy eosinófilo, que se sitúan alrededor de áreas de necrosis y de degeneración del colágeno con una disposición que recuerda a los granulomas «en empalizada»1,3,8,9. A menudo existe un infiltrado inflamatorio de predominio linfocitario que rodea a los nódulos tumorales, y pueden observarse zonas con áreas quísticas y hemorrágicas como resultado de la pérdida de cohesión celular y de necrosis, lo que origina un aspecto seudoangiomatoide2,3. En algunos de estos tumores se aprecian células rabdoides grandes, muy atípicas, con núcleo excéntrico y citoplasma globoide eosinófilo6,10. El estudio inmunohistoquímico es muy esclarecedor, aunque no decisivo, ya que suelen existir diferencias entre distintos tumores y aún dentro de un mismo tumor puede haber zonas donde varíen los resultados. Las citoqueratinas de bajo y alto peso molecular son positivas hasta en el 75 % de los casos, siendo en general más marcadas en las zonas donde predominan las células epitelioides que entre las fusiformes. El EMA y la vimentina son positivos hasta entre el 95 y el 100 % de los casos. En el 60-70 % de estos tumores existe positividad para CD-34, y este marcador es útil para los tumores que no son positivos para vimentina. La S-100, neurofilamentos, factor VIII y CD-31 son típicamente negativos2,10.
Se reconocen diversas variantes histopatológicas del sarcoma epitelioide. La variante angiomatoide muestra espacios quísticos hemorrágicos que recuerdan al angiosarcoma; en la forma similar a un fibroma predominan las células fusiformes con patrón estoriforme parecido al histiocitoma fibroso celular; la forma rabdoide está constituida por una sábana de grandes células rabdoides que simulan un tumor rabdoide de tejidos blandos y que suelen ser características del sarcoma epitelioide de localización proximal. Sin embargo, el estudio inmunohistoquímico de todas estas variantes es prácticamente similar10,11. Es probable que las variaciones en la celularidad estén en función del momento evolutivo en el que se estudia el tumor, con diferencias claras entre las formas primitivas y las recurrencias. Ya Enzinger1 indicó que las lesiones tempranas de sarcoma epitelioide tenían un mayor predominio de células fusiformes, mientras que las células epitelioides predominaban en las recurrencias y en las lesiones metastásicas. También la presencia de necrosis, de áreas angiosarcomatoides y de células rabdoides es más típica de lesiones recurrentes.
Los tumores primitivos muestran un aspecto fibromatoso y pueden confundirse con procesos fibrosos benignos de tipo reactivo12,13, como fascitis nodular, histiocitoma fibroso y fibromatosis2,7,10. Cuando son lesiones superficiales y están constituidas por uno o múltiples nódulos, se suele confundir con procesos inflamatorios de tipo granulomatoso como granulomas infecciosos, necrobiosis lipoídica, granuloma anular o nódulos reumatoides. En estadios más avanzados las células tumorales tienden a definirse más claramente, son más alargadas, más eosinófilas y adquieren hábito epitelioide. La positividad para citoqueratinas, EMA y vimentina, y la negatividad para otros muchos marcadores permiten realizar el diagnóstico. El sarcoma sinovial tiene las mismas características inmunohistoquímicas que el sarcoma epitelioide, pero se distingue de éste por el patrón seudoglandular o bifásico y la presencia de mucina intracelular2,7.
El tratamiento de elección de estos tumores es la extirpación quirúrgica radical con amplio margen en superficie y fascia muscular en profundidad en el caso de no estar ésta afectada, ya que sólo con grandes márgenes libres de tumor se podrá controlar el alto poder de recurrencias locales de este tumor. La amputación del miembro afectado es la actuación terapéutica más adecuada cuando el tumor primario está localizado en los dedos de la mano o el pie y en todos los casos de recidiva local del tumor primario2. A pesar de ello no está totalmente comprobado si este tipo de cirugía agresiva influye en la futura aparición de metástasis a distancia, que parece estar más relacionada con el tiempo de evolución de la enfermedad que con el tipo de tratamiento inicial3.
Se consideran tres variables anatomopatológicas que parecen influir en la supervivencia: la invasión vascular, que es la de mayor valor predictorio, el tamaño del tumor mayor de 5 cm y la presencia de más de un 30 % de necrosis3,9. No está aún establecido el beneficio del vaciamiento ganglionar regional, la quimioterapia y la radioterapia en la prevención de las recurrencias locales y de las metástasis a distancia, porque no existen series amplias que lo corroboren. De todas formas en los casos aislados en los que se ha utilizado esta terapia parece haber sido beneficiosa14,15. La mayoría de las recurrencias locales aparecen a los 2 años de la lesión inicial, aunque no son infrecuentes las recurrencias tardías3. Las áreas más frecuentes de metástasis son el pulmón, la pleura, los ganglios linfáticos y el cuero cabelludo5. Existen casos donde el tratamiento quirúrgico de las metástasis ganglionares y pulmonares ha prolongado la supervivencia3. La mortalidad del sarcoma epitelioide es del 27 % en los primeros 5 años de seguimiento3.



