

Rosai-Dorfman disease (RDD) is a rare, benign histiocytic disorder characterized by generalized lymphadenopathy and constitutional symptoms, secondary to histiocytic infiltration of lymph nodes. Cutaneous Rosai-Dorfman disease (CRDD), the variant limited to the skin, is very rare. We present a case of CRDD on the left cheek that responded well to methotrexate.
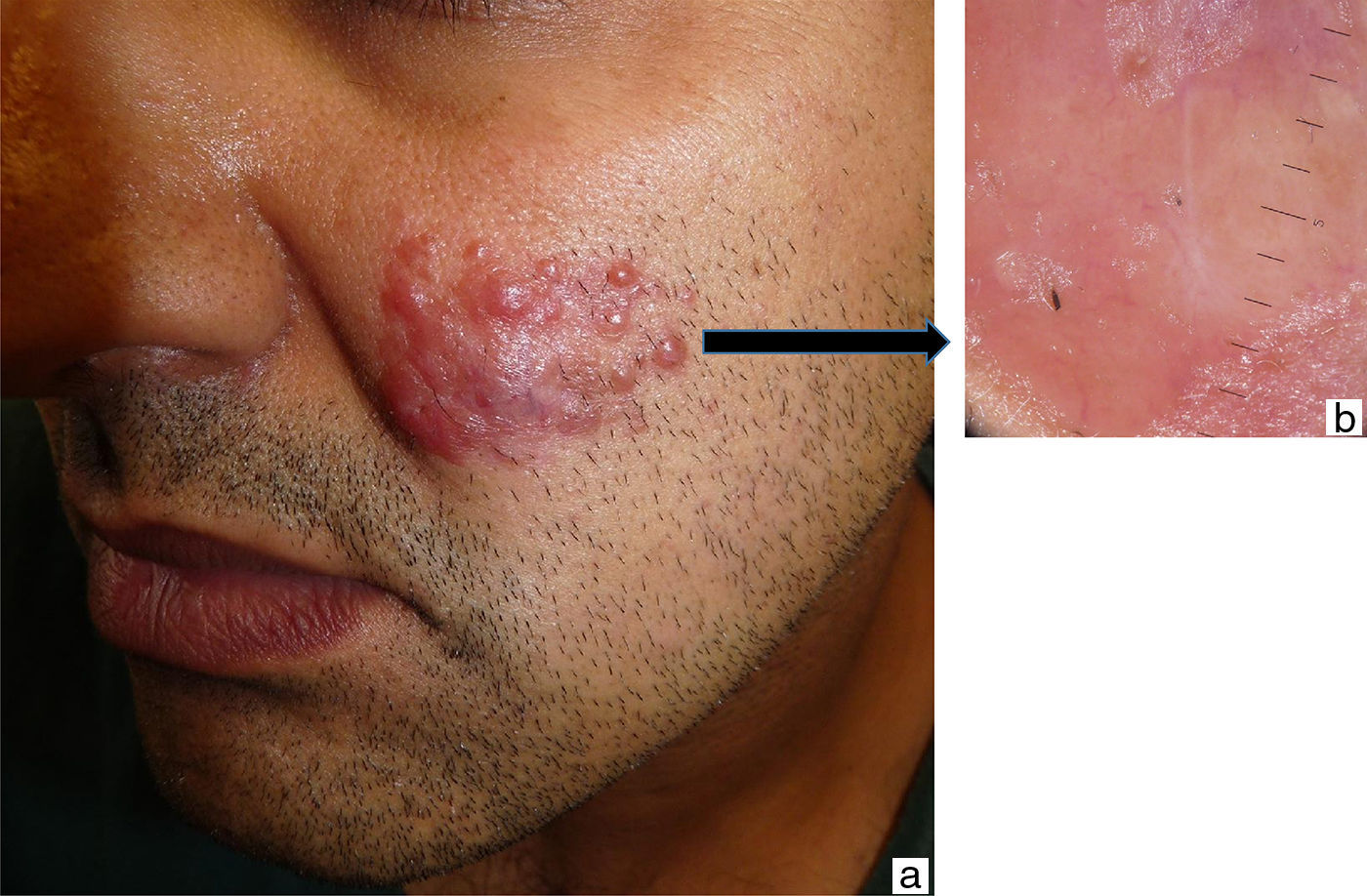
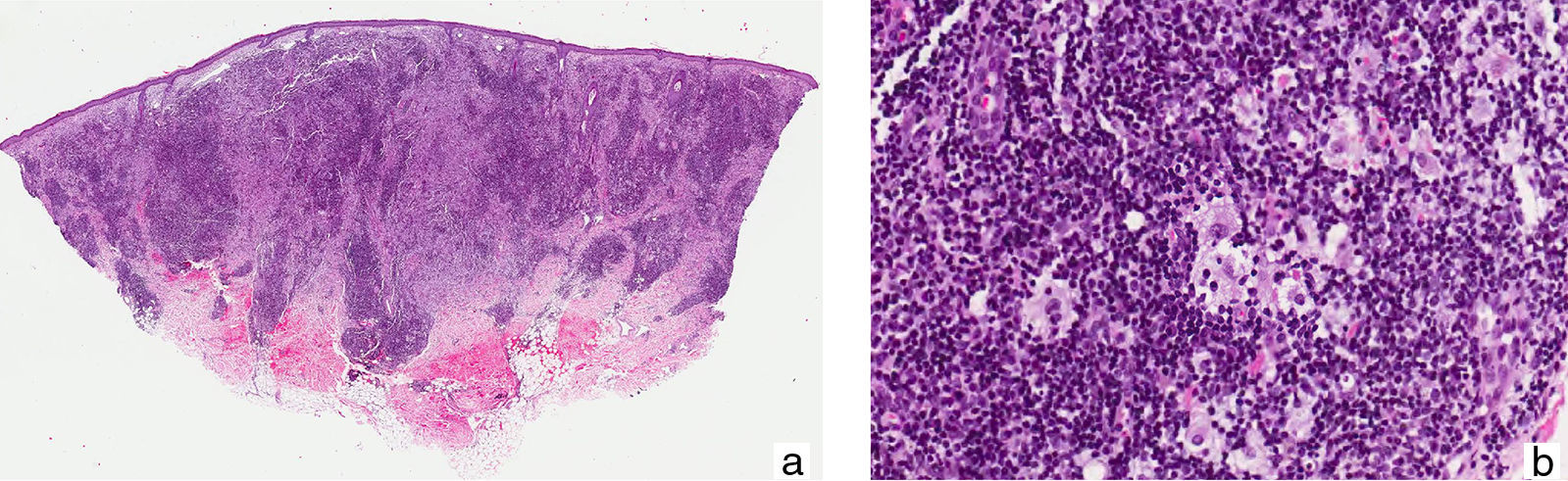
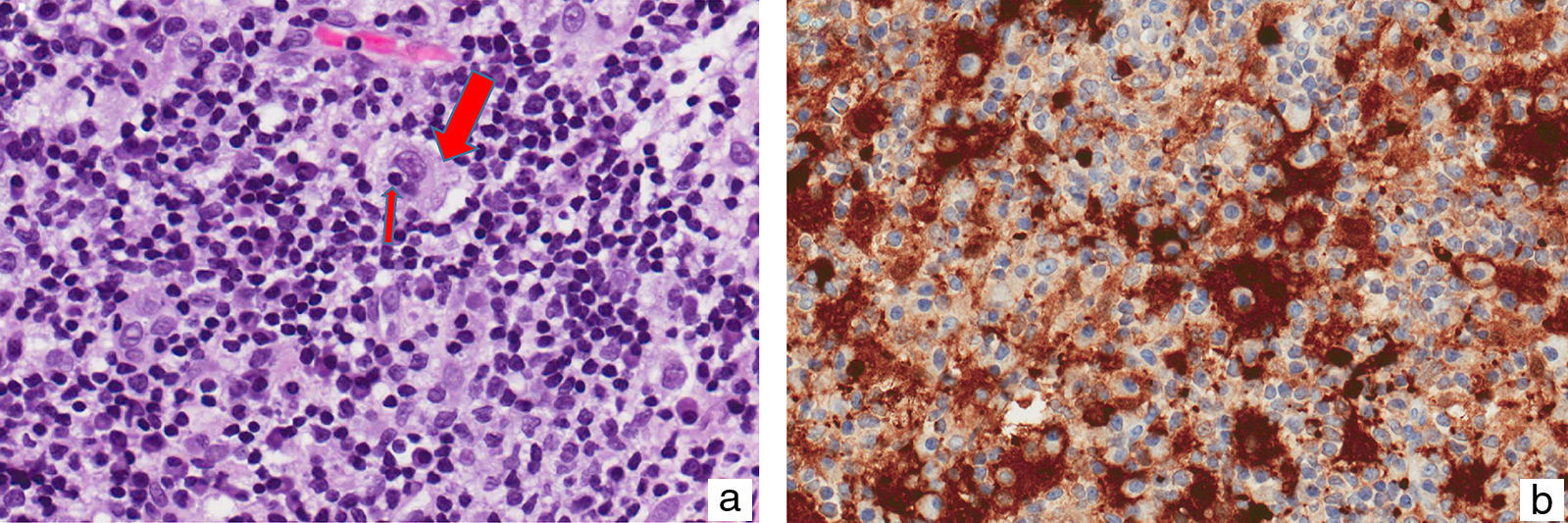
A 38-year-old Moroccan man who had been living in Spain for about 6 years presented with a slow-growing asymptomatic lesion on the left cheek that had first appeared 18 months earlier and was not associated with any other symptoms. Physical examination revealed nonulcerated, yellowish, erythematous papules and nodules that formed a 4cm infiltrated plaque (Fig. 1). The patient had no palpable lymph nodes or visceromegaly. Additional tests (complete blood count, biochemistry, erythrocyte sedimentation rate, C-reactive protein, angiotensin-converting enzyme, protein electrophoresis, immunoglobulins, complement, antinuclear antibodies, syphilis serology, hepatitis and human immunodeficiency virus (HIV), β2-microglobulin, and chest radiography) were normal or negative. Histopathologic examination revealed an unaffected epidermis and a granulomatous lymphohistiocytic inflammatory infiltrate (Fig. 2A) accompanied by plasma cells and few neutrophils in the dermis (Fig. 2B); histiocytes with abundant cytoplasm and vesicular nuclei showed striking phenomena of emperipolesis (intact inflammatory cells engulfed by histiocytes) (Fig. 3); and immunohistochemistry was positive for CD68 and S-100 protein and negative for CD1a. Microbiologic studies were negative for fungi, Mycobacterium tuberculosis, atypical mycobacteria and Leishmania spp. A diagnosis of CRDD was established on the basis of these findings and staging studies revealed no systemic involvement. Treatment was started with intralesional corticosteroids; partial response was achieved, so methotrexate (15mg/wk) was added. Clear improvement was observed at 2 months.

. B, Higher magnification shows an infiltrate composed of large histiocytes with vesicular nuclei, accompanied by abundant plasma cells and lymphocytes (hematoxylin-eosin, original magnification ×20).")
A, Granulomatous lymphohistiocytic infiltrate in the dermis (hematoxylin-eosin, original magnification ×10). B, Higher magnification shows an infiltrate composed of large histiocytes with vesicular nuclei, accompanied by abundant plasma cells and lymphocytes (hematoxylin-eosin, original magnification ×20).
 with lymphocytes in its cytoplasm (emperipolesis) (small arrow) (hematoxylin-eosin, original magnification ×40). B, Negative image of inflammatory cells compared to histiocytic cells with immunohistochemical staining for S-100 protein.")
RDD, or sinus histiocytosis with massive lymphadenopathy, is a form of non-Langerhans cell histiocytosis first described as a distinct entity in 1969 by Rosai and Dorfman1–4; it can occur in isolation or as part of other more complex conditions (R group histiocytoses).5 RDD mainly affects young white and African American men and usually manifests as bilateral cervical lymphadenopathy associated with fever, weight loss, night sweats, fatigue,5 leukocytosis with neutrophilia, and polyclonal hypergammaglobulinemia.6 It is sometimes associated with autoimmune disorders such as lupus erythematosus, autoimmune hemolytic anemia, Crohn disease, primary cutaneous marginal zone lymphoma with IgG4 expression, and HIV infection. Extranodal involvement is present in 25% to 40% of cases. The skin, affected in up to 10% of cases, is one of the most frequently involved organs.5,7 However, cases affecting only the skin are very rare.2 Just over 100 exclusively cutaneous cases have been reported,3,4,8 accounting for approximately 3% of all cases. Exclusively cutaneous cases most frequently affect middle-aged white and Asian women.
The clinical manifestations of CRDD are variable and nonspecific, including single or multiple papules, nodules, or plaques,4 or, less frequently, other presentations such as pustules, acneiform lesions, and lesions mimicking vasculitis and panniculitis.3 The face is the most common site, followed by the back, chest, thighs, hips, and shoulders. The presence of reddish-yellow nodules without tenderness to palpation can be useful in establishing a diagnosis.3
Histologically, the epidermis shows no abnormalities and a diffuse inflammatory infiltrate of histiocytes accompanied by lymphocytes, numerous plasma cells,5 and isolated neutrophils is observed in the dermis. Phenomena of emperipolesis, an essential—although not pathognomonic—feature for diagnosis,1 indicate that intact inflammatory cells and/or erythrocytes are present in intracytoplasmic vacuoles inside histiocytes, allowing them to be spared degradation by cytolytic enzymes,1,3 in contrast to phagocytosis, in which the cells are destroyed. Nuclear atypia and mitotic figures are rare. Histiocytes are positive for S-100 protein and CD68 and negative for CD1a, which helps confirm the diagnosis and rule out other entities,3 especially in extranodal lesions, which present a much lower frequency of emperipolesis.1
The etiology of CRDD is unknown1 but it has been suggested that a viral infectious agent—for example, herpesvirus, Epstein-Barr virus, or parvovirus B19—and genetic factors could play a role. It has also been hypothesized that CRDD is an inflammatory disorder2 because the polyclonal nature of the infiltrate suggests a reactive rather than neoplastic process.3 Unlike Langerhans cell histiocytosis, no BRAFV600E mutations have been detected in the studied cases, as in other histiocytic disorders such as juvenile xanthogranuloma.
The prognosis of CRDD is generally favorable2,4 and many cases resolve spontaneously.7 Numerous treatments—including topical and systemic corticosteroids, thalidomide, dapsone, retinoids, cryotherapy, and radiation therapy3—have been used, all with variable efficacy. In refractory cases, vincristine9 and imatinib10 have shown very good results. A recent case was treated with low-dose methotrexate, with good response.4 Surgical removal can be justified in localized cases.
We present a new case of facial CRDD; it is important to consider this entity in the differential diagnosis of facial lesions with a granulomatous appearance in order to avoid diagnostic delays, so that treatment can be started if necessary.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
The authors would like to thank Dr. Santiago Montes and the team of Dr. M.A. Piris from Hospital Universitario Marqués de Valdecilla (Santander) for their collaboration on the diagnosis.
Please cite this article as: Flores-Terry MÁ, Romero-Aguilera G, González-López L, García-Arpa M. Enfermedad de Rosai-Dorfman cutánea facial. Actas Dermosifiliogr. 2017;108:479–481.



