

A recent event of considerable significance for the pharmaceutical industry may have been overlooked by many dermatologists in spite of its great relevance to our specialty: I refer to the approval of Remsima by South Korea's Food and Drug Administration (FDA) on July 23, 2012. Remsima (previously, CT-P13) is the first biosimilar version of a monoclonal antibody (specifically infliximab, or Remicade).1 Although Reditux, a rituximab (MabThera) biosimilar, was approved in India in 2007, the Indian standards differ considerably from international ones, whereas the Korean standards do not.
Celltrion, the laboratory that developed CT-P13 and has invested some USD $200 million in Remsima so far,1 brought the results of the PLANET trials to the EULAR congress in Berlin earlier in 2012. Those trials, in which 125 centers in 19 different countries participated, included a phase I trial in 250 patients with ankylosing spondylitis and a phase III trial in 580 patients with rheumatoid arthritis.2 The Korean firm has also applied to the European Medicines Agency (EMA) for approval of the new drug and plans to market it with Hospira3 starting in 2014, when patents for Remicade expire in the European Union. Applications have also been filed in Asia, Central America, and South America, where Remicade is not under patent protection. Whether Celltrion has included other uses for Remicade in the applications is still unknown, given that EU standards stipulate that once comparability has been demonstrated, indications can be extrapolated if “adequately justified”–although adequacy is judged on a case-by-case basis. The EMA has also announced that another application for an infliximab biosimilar has been filed,4 although the name of the laboratory has not been revealed.
Without doubt, the starting pistol has been fired in a race between many competing laboratories in 3 geographic areas- Europe, the Far East, and India-as companies vie for the largest share of this particular pharmaceutical industry pie. Savings from widespread use of generics and biosimilars worldwide are potentially enormous. This interest along with the profit motive explains the large number of contenders jockeying for position even though their individual shares must remain small in the short term. Public health systems partially fund the development of biosimilars in countries like Korea and Singapore, and the authorities in Indonesia and Thailand are considering joining them provided quality, safety, and price are acceptable and the drugs target local health priorities. Countries like Korea and Japan, with well-established industrial and pharmaceutical capabilities, have also redirected industrial efforts to biotechnology. Something similar is happening in China and in emerging economies like India, Brazil, and Mexico, where some pharmaceutical companies have become strong producers of generics. An example is Dr Reddy's Laboratories, an Indian company that launched Reditux, a biosimilar that sells at half the price of the originator molecule and has increased the Indian market for rituximab treatment 10-fold.5 A June 2012 agreement between Dr. Reddy's and Merck Serono will allow the Indian company to sell its product in Europe,6 and the Indian producer has made similar agreements with GlaxoSmithKline for other biosimilars.3
When biosimilars enter the market, the expected benefits include lower costs that will make treatments more accessible to more patients and sustainable for health care systems. The main limitations–apart from possible problems with technology, efficacy or safety, and regulatory issues-will stem from tough legal battles that the owners of patents still in force can be expected to wage. The biosimilars industry accounts for 20% of research and development spending in the United States. In 2010, monoclonal antibodies alone made up a market segment amounting to USD $46 billion, and annual growth of 12% is expected.7 This sector therefore has strategic importance for US export policies and generates significant employment.
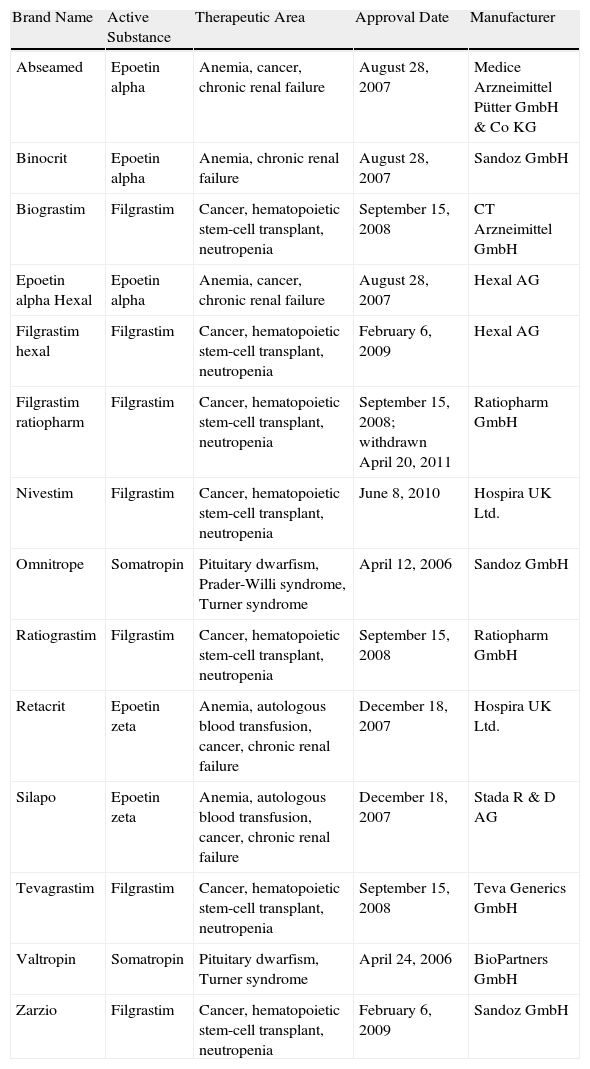
The biosimilars that have passed the EU approval process to date (Table 1) include erythropoietins, granulocyte colony-stimulating factors, and somatropin; all these products have molecular weights 10 times lower than those of monoclonal antibodies, which are considered more complex molecules to produce and copy. Although the manufacture of biosimilars is now simpler and less costly, it remains complex.8 Proteins are produced by inserting a fragment of DNA into a cloning vector for transfer to a selected cell line (such as Chinese hamster ovary cells) that is expanded in fermentation tanks (bioreactors). In principle, a complex purification, validation, and quality control process ensures product stability. However, the amino acid sequence is not the only concern in more complex molecules, such as immunoglobulins (with molecular weights of around 160kDa). Other relevant characteristics that affect the activity and immunogenicity of the biological product include the molecule's tridimensional conformation or quaternary structure, in which methylation or glycosylation of certain residues intervene, as well as aggregation and the presence of impurities.9
Biosimilars Approved by the European Medicines Agency as of July 1, 2012.a
| Brand Name | Active Substance | Therapeutic Area | Approval Date | Manufacturer |
| Abseamed | Epoetin alpha | Anemia, cancer, chronic renal failure | August 28, 2007 | Medice Arzneimittel Pütter GmbH & Co KG |
| Binocrit | Epoetin alpha | Anemia, chronic renal failure | August 28, 2007 | Sandoz GmbH |
| Biograstim | Filgrastim | Cancer, hematopoietic stem-cell transplant, neutropenia | September 15, 2008 | CT Arzneimittel GmbH |
| Epoetin alpha Hexal | Epoetin alpha | Anemia, cancer, chronic renal failure | August 28, 2007 | Hexal AG |
| Filgrastim hexal | Filgrastim | Cancer, hematopoietic stem-cell transplant, neutropenia | February 6, 2009 | Hexal AG |
| Filgrastim ratiopharm | Filgrastim | Cancer, hematopoietic stem-cell transplant, neutropenia | September 15, 2008; withdrawn April 20, 2011 | Ratiopharm GmbH |
| Nivestim | Filgrastim | Cancer, hematopoietic stem-cell transplant, neutropenia | June 8, 2010 | Hospira UK Ltd. |
| Omnitrope | Somatropin | Pituitary dwarfism, Prader-Willi syndrome, Turner syndrome | April 12, 2006 | Sandoz GmbH |
| Ratiograstim | Filgrastim | Cancer, hematopoietic stem-cell transplant, neutropenia | September 15, 2008 | Ratiopharm GmbH |
| Retacrit | Epoetin zeta | Anemia, autologous blood transfusion, cancer, chronic renal failure | December 18, 2007 | Hospira UK Ltd. |
| Silapo | Epoetin zeta | Anemia, autologous blood transfusion, cancer, chronic renal failure | December 18, 2007 | Stada R & D AG |
| Tevagrastim | Filgrastim | Cancer, hematopoietic stem-cell transplant, neutropenia | September 15, 2008 | Teva Generics GmbH |
| Valtropin | Somatropin | Pituitary dwarfism, Turner syndrome | April 24, 2006 | BioPartners GmbH |
| Zarzio | Filgrastim | Cancer, hematopoietic stem-cell transplant, neutropenia | February 6, 2009 | Sandoz GmbH |
Because the manufacturers of biosimilars lack access to information about the production process of the patented originator, it is very difficult for them to replicate the glycosylation pattern of the innovator molecule. According to a proposal drafted in the United States, however, the original producer can gain access to the documents a competitor files to support an application for approval of a biosimilar. In this situation, it is impossible to manufacture an identical molecule unless industrial secrecy is violated or the original laboratory decides to create a copy of its own drug, changing the delivery device for example; rather, what can be developed is a biologically and clinically comparable molecule. Such drugs have been termed biosimilars in the EU, follow-on biologics in the United States, and subsequent entry biologics in Canada.
In developed countries, the approval processes for biosimilar drugs and generics differ. Generics are small, well-defined, chemically synthesized molecules and only their pharmacokinetic comparability must be demonstrated; moreover, as a certain range of comparability is tolerated, a generic might only meet requirements at the lower end of that range. Biosimilars, on the other hand, require additional studies beyond the demonstration of chemical similarity. Comparative pharmacokinetic and pharmacodynamic trials in healthy volunteers or patients must be undertaken at different phases, and if the originator molecule has been derived from more than a single biologic source, comparing all of them entails higher clinical trial costs. At least 1 clinical trial is required to demonstrate the equivalence (or noninferiority, which requires fewer patients and lowers costs) of each available formulation (for subcutaneous or intravenous administration, for instance).8
An equivalence trial must include a definition of the minimal clinically relevant difference between 2 treatments with regard to a measure of efficacy; in comparing response rates, a maximum allowable difference (Δ) on which to base an assumption of equivalence between a biosimilar and the innovator molecule has been considered to be 15%. As Δ increases, the sample required to test a hypothesis decreases; for example, if Δ is reduced by half, the required sample size increases 4-fold. The sample size also decreases when the numerical value of an outcome measure is greater; so for a given Δ, it will be cheaper to demonstrate the clinical comparability of a biosimilar if the efficacy endpoint is a 50% improvement in the psoriasis area severity index rather than a 75% improvement. It is important to remember that clinical trials are the driver in drug development costs, while production costs are marginal even for biologics. Thus, simplifying the clinical development process can make biosimilar drugs more affordable.
A biosimilar product's efficacy can be extrapolated to other indications previously approved for the innovator drug when the pathogenic mechanisms are similar, with no need for independent clinical trials. However, the final decision rests with the regulatory authorities, who will rule on a case-by-case basis. All trials must evaluate the product's immunogenicity and safety and a pharmacovigilance program must have been designed and be ready for implementation following the product's approval.
The latest version of EMA regulations (effective, December 1, 2012)10 is silent on the matter of allowing biosimilar products to be switched, or interchanged, for the originator drugs: that decision is actually the responsibility of each EU member state. In contrast, a generic can be freely interchanged for an original drug. US legislation (US Biologics Price Competition and Innovation Act11), on the other hand, allows the FDA to designate biosimilars that can be switched at the pharmacist's discretion without consulting the prescriber. Although switching biologics to biosimilars has been criticized for the reasons discussed above, in fact originator products themselves change over time,12 particularly when manufacturing processes are modified. For this reason, it is recommended that some leeway be established when setting limits on acceptable comparability for biosimilars with respect to the originators13; strictly speaking, the original manufacturer is actually producing a biosimilar of its own drug with each new batch or change in the manufacturing process. Therefore, rather than demonstrating maximum similarity to the originator, biosimilar drug producers should demonstrate that theirs is as similar to the originator as are the products in the various batches produced throughout the drug's lifetime on the market.14
In fact, changes in formulation can cause adverse events: when human serum albumin was replaced with polysorbate 80 and glycine (as required by regulatory authorities) as stabilizers for epoetin alpha at the end of the 1990s, numerous cases of pure red cell aplasia occurred due to cross reactivity between circulating erythropoietin and antibodies that formed in response to the biologic.15 Although such events are highly improbable when a product consists of an antibody against a pathogenic target rather than when it replaces a molecule with a physiological function, this outbreak has been a cautionary tale that both regulatory agencies and specialist prescribers take very seriously.
At present, the EMA has issued clear directives16 and an abbreviated approval procedure for biosimilars, and the frameworks constructed by regulatory authorities in Canada, Japan, and Australia, for example, are similar. The ongoing debate on this point has been heated in the United States, and although an abbreviated pathway–known as the 351(k) application process–has been laid out for biosimilars there too, definitive rules have yet to be set down. Eventual producers of biosimilars can also become targets in fierce legal battles over patents in that country, and in consequence, even biosimilar manufacturers will probably ignore the abbreviated procedures and instead follow the traditional pathway for biologics–section 351(a) of the Biologic License Application–in order to avoid having to publish manufacturing procedures and specifications and to reduce their vulnerability to litigation.17 In India, China, and some countries in South and Central America, regulation is not at all similar to that of the aforementioned countries and can be described as less stringent. “Alternative” biologics have therefore been approved in these countries even though they have marked chemical, pharmacokinetic, pharmacodynamic or clinical differences with respect to the originator molecule. Until the specifications for these biosimilars can meet the quality, purity, bioequivalence, immunogenicity, and safety standards of the EMA, the USFDA, or similar regulators, they circulate only in unregulated or semiregulated markets.
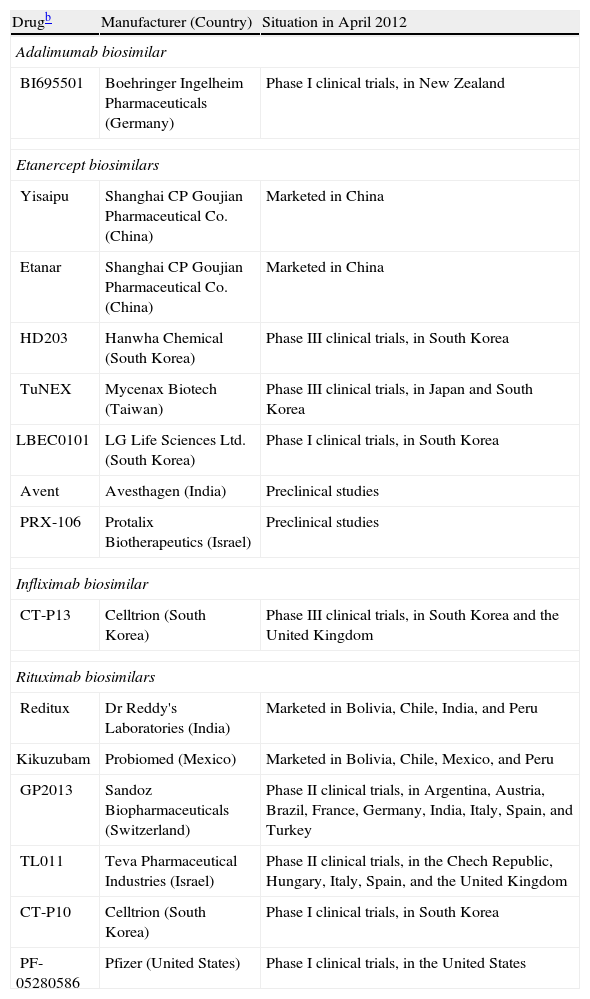
For many reasons, we can expect to see the development of biosimilars for the biologics used to treat psoriasis (Table 2),18 at least outside the United States, where current legislation protects innovator biologics for 12 years after FDA approval regardless of when the patent was filed.19
Biosimilars Presently on the Market or Under Development for Treating Rheumatic Diseases and Psoriasis.a
| Drugb | Manufacturer (Country) | Situation in April 2012 |
| Adalimumab biosimilar | ||
| BI695501 | Boehringer Ingelheim Pharmaceuticals (Germany) | Phase I clinical trials, in New Zealand |
| Etanercept biosimilars | ||
| Yisaipu | Shanghai CP Goujian Pharmaceutical Co. (China) | Marketed in China |
| Etanar | Shanghai CP Goujian Pharmaceutical Co. (China) | Marketed in China |
| HD203 | Hanwha Chemical (South Korea) | Phase III clinical trials, in South Korea |
| TuNEX | Mycenax Biotech (Taiwan) | Phase III clinical trials, in Japan and South Korea |
| LBEC0101 | LG Life Sciences Ltd. (South Korea) | Phase I clinical trials, in South Korea |
| Avent | Avesthagen (India) | Preclinical studies |
| PRX-106 | Protalix Biotherapeutics (Israel) | Preclinical studies |
| Infliximab biosimilar | ||
| CT-P13 | Celltrion (South Korea) | Phase III clinical trials, in South Korea and the United Kingdom |
| Rituximab biosimilars | ||
| Reditux | Dr Reddy's Laboratories (India) | Marketed in Bolivia, Chile, India, and Peru |
| Kikuzubam | Probiomed (Mexico) | Marketed in Bolivia, Chile, Mexico, and Peru |
| GP2013 | Sandoz Biopharmaceuticals (Switzerland) | Phase II clinical trials, in Argentina, Austria, Brazil, France, Germany, India, Italy, Spain, and Turkey |
| TL011 | Teva Pharmaceutical Industries (Israel) | Phase II clinical trials, in the Chech Republic, Hungary, Italy, Spain, and the United Kingdom |
| CT-P10 | Celltrion (South Korea) | Phase I clinical trials, in South Korea |
| PF-05280586 | Pfizer (United States) | Phase I clinical trials, in the United States |
Rituximab is the monoclonal antibody that has been with us the longest and several companies already market biosimilars or have products in advanced stages of clinical development.
As for the fusion protein etanercept, Yisaipu and Etanar, produced by Shanghai CP Goujian Pharmaceutical Co, are equivalent products, sold in China and Colombia, respectively; the etanercept biosimilar Qiangke, produced by competitor Shanghai Celgen in partnership with Simcere Pharmaceuticals, was recently approved in China.20 The Korean laboratory Hanwha has registered its biosimilar (HD203) with South Korean regulators and is planning to cooperate with Merck on the eventual distribution of the drug outside the United States.21 The Japanese laboratory Daiichi Sankyo has come to an agreement with Coherus BioSciences to manufacture and distribute etanercept and rituximab biosimilars developed by the latter company,22 which specializes in biosimilars and has its headquarters in the United States. Once the new biosimilars are approved they will be sold through Daiichi Sankyo in Japan, South Korea, and Taiwan.
The approval of the Remicade biosimilar Remsima was mentioned at the beginning of this article, and Boehringer Ingelheim's adalimumab biosimilar (BI695501) is in the clinical development phase.23 PharmaPraxis plans to develop a Humira biosimilar in Brazil,24 and Fujifilm Kyowa Kirin Biologics (a joint venture of the giant holding company Fujifilm and a Japanese biotechnology company) has plans for biosimilars of adalimumab and other biologics.25
The patents for Remicade, Enbrel, and Humira will expire in most EU countries in 2014 (August 13), 2015 (February 1), and 2018 (April 16), respectively.26 The situation is different in the United States. The patent for Humira (whose worldwide sales amounted to USD $7.9 billion last year27) expires on December 31, 2016, in spite of recent extensions.28 However, Abbot Laboratories has filed a “citizen petition” with the FDA asking the agency to decline to review any application for a Humira biosimilar filed before March 23, 2010, when the Biologic Price Competition and Innovation Act entered into effect to regulate the evaluation procedures. The petitioners argue that approval of such applications would endanger industrial secrets protected under the Fifth Amendment to the US Constitution.29 The patent for Remicade is set to expire on September 4, 2018–20 years after it took effect. The case of Enbrel is also interesting. The US patent for this drug expires on October 23, 2012, but US Patent 8,063,182 was issued to Roche for it at the beginning of this year. The license for this molecule was ceded to Amgen (which acquired the previous licensee, Immunex, in 2002) after a one-time payment in 2004. Thus, the history of the patent filed in 1990 has been tortuous (or perhaps overtly twisted), but acceptance of the new filing gives Amgen 16 more years of protection for Enbrel in the United States.30
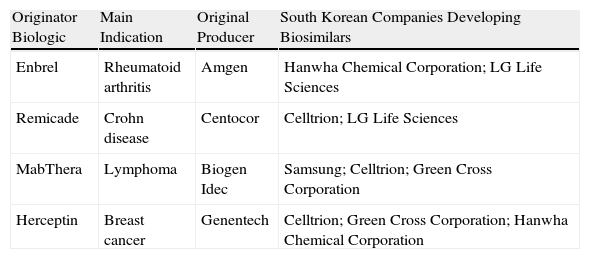
In direct contrast with the litigious environment in the US pharmaceuticals industry, in Korea a cluster of high-tech industries with innovative capabilities (Table 3)31 has emerged through joint ventures with giant industrial conglomerates such as Samsung Electronics.32 These large enterprises are redefining the strategic importance of the pharmaceutical sector. Similar developments are occurring in Japan. As for China, a few years ago the production of biosimilars was based on roller bottles and 100-L bioreactors in many facilities, but manufacturing processes are now being brought up to standard and the presence of China at large conferences bears witness to the vitality of the sector in that country.33 Regardless of the short-term market impact of companies’ maneuvering, the boom in biosimilars will probably have played itself out in about 10 years, partly because the market will be saturated but also because patents for new innovator drugs will not yet be approaching their patent expiry dates.
Capability in South Korea.
| Originator Biologic | Main Indication | Original Producer | South Korean Companies Developing Biosimilars |
| Enbrel | Rheumatoid arthritis | Amgen | Hanwha Chemical Corporation; LG Life Sciences |
| Remicade | Crohn disease | Centocor | Celltrion; LG Life Sciences |
| MabThera | Lymphoma | Biogen Idec | Samsung; Celltrion; Green Cross Corporation |
| Herceptin | Breast cancer | Genentech | Celltrion; Green Cross Corporation; Hanwha Chemical Corporation |
By then, biologics and biosimilars, with their many advantages of efficacy and safety, will have become much more affordable for patients and health care systems, and a powerful, newly-minted industry with advanced research and development capabilities and innovative production methods will have become well established.
We in Spain can also hope to catch the wave-even if we are not world-class surfers and do not find jet skis affordable. At the very least, we can hope to be carried along in its wake rather than remain at the farthest edges in the second quarter of this century.
Conflicts of InterestDr Lluis Puig has received consultancy and/or speaking fees or has participated in clinical trials funded by Abbott, Amgen, Celgene, Janssen, Merck Serono, MSD, Novartis, and Pfizer. All information in this article derives from sources that are freely available online or are published in journals indexed in PubMed.
Please cite this article as: Puig L. Biosimilares en dermatología: infliximab para empezar. Actas Dermosifiliogr. 2013;104:175–80.



