


Research into molecular targets for drug development in melanoma is starting to bear fruit. Of the drugs tested to date in patients with metastatic melanoma, those that have yielded the best results are V600E BRAF inhibitors in melanomas carrying the V600E mutation; c-kit tyrosine kinase activity inhibitors in melanomas carrying c-kit mutations; and anti-cytotoxic T lymphocyte antigen 4 (CTLA-4) antibodies, which block the mechanisms involved in immune tolerance. Many problems have yet to be resolved in these areas, however, such as the rapid development of resistance to BRAF and c-kit inhibitors and the lack of biomarkers to predict treatment response in the case of CTLA-4 blockers. We review the results of targeted therapy with these and other drugs in metastatic melanoma and discuss what the future holds for this field.
La investigación sobre dianas moleculares en el melanoma sobre las que se pueda actuar farmacológicamente está empezando a dar sus primeros frutos. De todos los fármacos ensayados hasta el momento en pacientes con melanoma diseminado, los que han conseguido mejores resultados son los inhibidores de la mutación V600E de BRAF en los melanomas portadores de la misma, los inhibidores de la actividad tirosin-cinasa de c-Kit en melanomas con mutaciones de este gen y los anticuerpos anti-CTLA-4, inhibidores de los mecanismos de inmunotolerancia. Sin embargo, aún quedan muchos problemas por resolver, como la rápida adquisición de resistencias frente a los dos primeros tipos de fármacos o la falta de biomarcadores predictivos de respuesta frente al último de ellos. En este artículo presentamos una revisión sobre los resultados de los tratamientos contra estas y otras dianas en el melanoma diseminado y lo que parece que podemos esperar del futuro.
Once cutaneous melanoma has metastasized, it is extremely refractory to conventional antineoplastic treatments. At present, dacarbazine is still the standard treatment for patients with metastatic melanoma. The objective response rate is just 10% to 20% while the complete response rate is less than 5% and lasts only 6 to 8 months. Therefore, much effort is being put into developing new therapeutic strategies.1,2 New therapies directed against molecular targets have revolutionized other areas of oncology3 but until fairly recently such approaches were not thought to be viable in metastatic melanoma. However, results just published are providing some encouragement.
Targeted treatments act by selectively inhibiting molecules, usually proteins, whose expression or overexpression plays a specific role in the growth of the target neoplasm. Thus, one of the main characteristics of targeted antineoplastic therapy is that the drugs act specifically on their intended target and that those targets have specific effects on the tumor. The advantage of this specificity is that the treatment avoids the toxicity of conventional antineoplastic therapy with nonspecific effects on malign and normal cells alike. Another advantage is that it is possible to discriminate between groups of patients that are apparently similar. One example now fully incorporated into routine clinical practice is the indication of trastuzumab for the treatment of breast cancer only when the human epidermal growth factor receptor (HER) 2 protein is overexpressed.4 Although such specificity is not always achieved and, as dermatologists, we are witness to many of the side effects of these new drugs,5 the fact remains that increasingly specific treatments are available for each type and subtype of cancer, allowing tailored therapies.The number of possible therapeutic targets in melanoma is increasing as our understanding of the biology of this tumor improves. Different drugs have been synthesized to target some of the molecules implicated in favoring tumor growth (Table 1).6–8
Potential Target Therapies in Melanoma.
| Targets within the tumor cells |
| Targeting molecules responsible for stimulating growth and/or preventing cell death |
| - Drugs that target antiapoptotic molecules (oblimersen, ABT-737, ABT-263, YM155) |
| - Inhibitors of proteins in the MAP kinase cascade: |
| • Inhibitors of farnesyl transferase (target RAS) (tipifarnib) |
| • RAF inhibitors (sorafenib, PLX4032, GSK2118436, RAF-265, XL281) |
| • MEK inhibitors (PD0325901, AZD6244, GSK1120212, E6201) |
| - Inhibitors of the PI3K/AKT pathway: |
| • Rapamycin analogs (target mTOR) (temsirolimus/CCI-779) |
| • Dual inhibitors of PI3K and mTOR (SF-1126, NVP-BEZ235, NVP-BGT226, XL765) |
| • PI3K inhibitors (PX-866, XL147, NVP-BKM120, GDC-0941, CAL-101) |
| • AKT inhibitors (MK-2206, GSK690693) |
| - c-kit inhibitors (imatinib, sunitinib, dasatinib, nilotinib) |
| - Inhibitors with pleiotropic action: |
| • Proteasome inhibitors (bortezomib and second-generation inhibitors) |
| • Histone deacetylase inhibitors (vorinostat and others) |
| Targeting molecules responsible for facilitating invasion and/or metastasis |
| - Antiadhesion molecule therapy: |
| • Anti-integrin antibodies αvβ3 (etaracizumab) |
| • Caderin N inhibitors (ADH-1) |
| • Anti-MCAM/MUC18 antibodies (ABX-MA1) |
| Targets in structures other than neoplastic cells |
| Antiangiogenic drugs |
| - Anti-VEGF antibodies (bevacizumab) |
| - VEGFR inhibitors (sunitinib, sorafenib, semaxanib, axitinib) |
| - Integrin antagonists (cilengitide) |
| - Thalidomide and derivatives (lenalidomide) |
| Inhibitors of immune tolerance mechanisms |
| - Anti-CTLA-4 antibodies (ipilimumab and tremelimumab) |
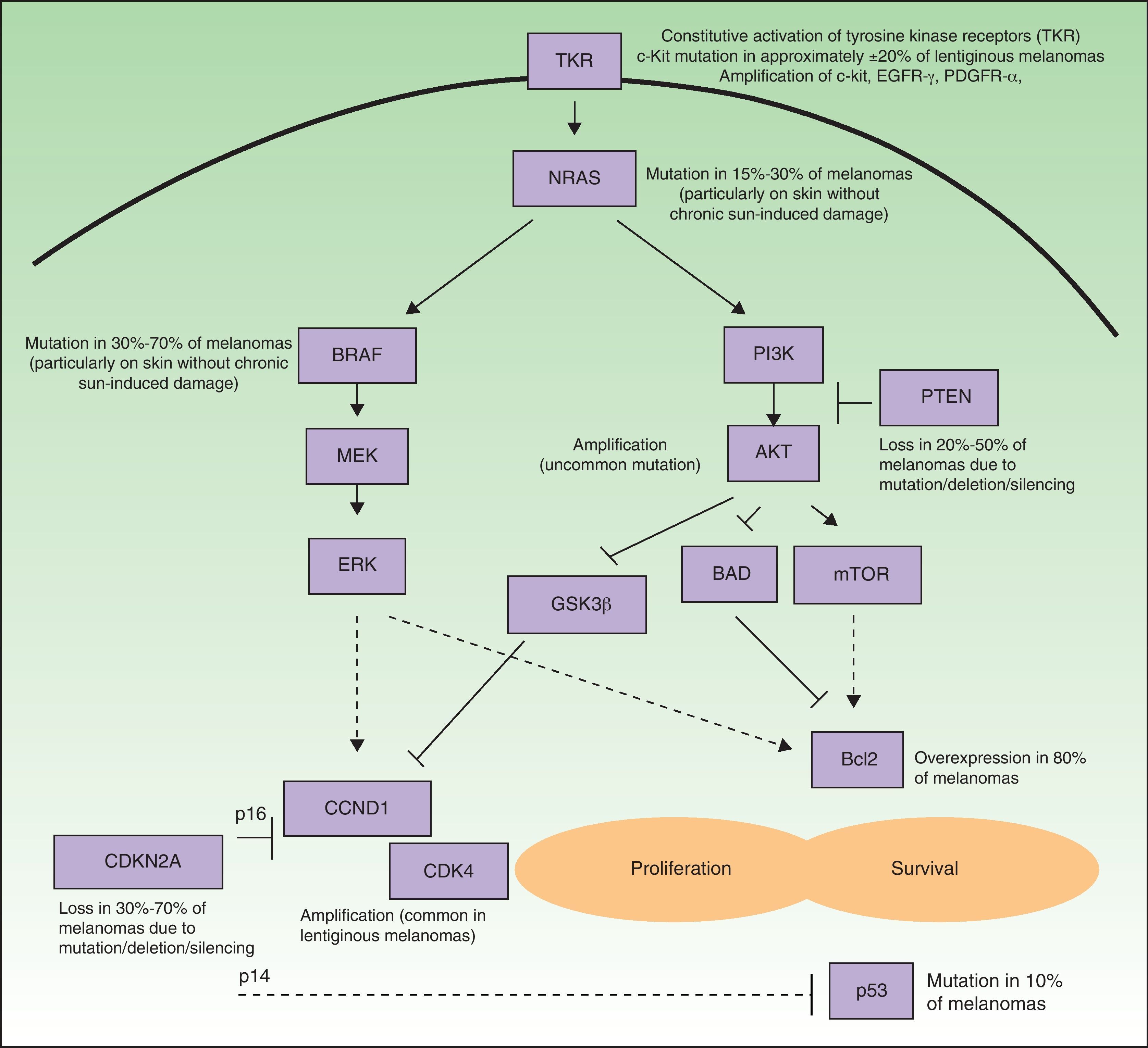
Many of the targets within tumor cells are growth factors, growth factor receptors, and proteins implicated in intracellular signaling pathways with proproliferative or antiapoptotic effects, and most are products of oncogenes.6 Such pathways in melanoma cells are highly activated (Fig. 1), either through mutations in the genes that encode proteins implicated in these pathways or through variations in levels of protein expression. As a result, the cells have a high proliferative capacity and a natural resistance to the extrinsic and/or intrinsic mechanisms that induce programmed cell death or apoptosis. Some of the mechanisms responsible for these abnormal cell signaling networks are as follows: 1) constitutive activation of growth factor receptors (c-kit, platelet-derived growth factor [PDGFR] α, epidermal growth factor receptor [EGFR]); 2) activation of the signaling pathways of mitogen-activated protein [MAP]-kinases (RAS/RAF/MAP kinase kinase [MEK]/extracellular signal-regulated kinase [ERK]); 3) constitutive activation of the AKT pathway (phosphatidylinositol 3-kinase [PI3K]/AKT), enhanced among other mechanisms by, for example, mutations, deletions, or silencing of the phosphatase and tensin homolog (PTEN) tumor suppressor gene; 4) alterations in the cell cycle control network (deletion, silencing, or mutation of cyclin-dependent kinase inhibitor [CDKN] 2A, amplification of cyclin-dependent kinase [CDK] 4, or cyclin D [CCND] 1); 5) deterioration of the transcriptional activity of proapoptotic protein p53; and 6) overexpression of antiapoptotic proteins of the Bcl-2 family as a result of aberrations in several of the aforementioned intracellular signaling pathways.9–11 Any of the proteins located at strategic points in these pathways could in principle be a good molecular target for the treatment of metastatic melanoma.
 and survival (stimulation of antiapoptic mechanisms and suppression of the proapoptotic mechanisms). Many melanomas have constitutive activation of growth factor receptors with tyrosine-kinase activity (TKR) (for example, c-kit, PDGFR-α, and/or EGFR), whether through amplifications or mutations of the genes that encode them. Likewise, they show activation of the signaling pathways of the mitogen-activated protein (MAP) kinases (RAS/RAF/MEK/ERK), due essentially to NRAS and BRAF mutations. Often the phosphatidyl-inositol 3 kinase (PI3K) (PI3K/AKT) pathway is also activated indirectly as a result of NRAS mutations and AKT amplification, and is especially favored by the loss of the inhibitory role of PTEN, resulting from mutations, deletions, or silencing of the tumor suppressor gene PTEN. In addition, deletions, silencing, and mutations of CDKN2A are implicated in activation of the cell cycle. These genetic aberrations lead to defects in the 2 proteins encoded by this gene (p16 and p14), as well as in CDK4 and CCND1 (cyclin D1) amplifications. This all has a deleterious impact on the activity of the proapoptotic protein p53 (which may also be mutated in some cases) and overexpression of antiapoptotic proteins of the Bcl-2 family.")
Simplified diagram of the some of the main intracellular pathways implicated in enhancing melanoma cell proliferation (stimulation of the cell cycle) and survival (stimulation of antiapoptic mechanisms and suppression of the proapoptotic mechanisms). Many melanomas have constitutive activation of growth factor receptors with tyrosine-kinase activity (TKR) (for example, c-kit, PDGFR-α, and/or EGFR), whether through amplifications or mutations of the genes that encode them. Likewise, they show activation of the signaling pathways of the mitogen-activated protein (MAP) kinases (RAS/RAF/MEK/ERK), due essentially to NRAS and BRAF mutations. Often the phosphatidyl-inositol 3 kinase (PI3K) (PI3K/AKT) pathway is also activated indirectly as a result of NRAS mutations and AKT amplification, and is especially favored by the loss of the inhibitory role of PTEN, resulting from mutations, deletions, or silencing of the tumor suppressor gene PTEN. In addition, deletions, silencing, and mutations of CDKN2A are implicated in activation of the cell cycle. These genetic aberrations lead to defects in the 2 proteins encoded by this gene (p16 and p14), as well as in CDK4 and CCND1 (cyclin D1) amplifications. This all has a deleterious impact on the activity of the proapoptotic protein p53 (which may also be mutated in some cases) and overexpression of antiapoptotic proteins of the Bcl-2 family.
The important realization that melanoma is genetically heterogeneous, whereby the aforementioned abnormalities may vary according to the subgroup of melanoma, has changed the approach to treatment of metastatic melanoma. Although previous data were already pointing along these lines, the first really solid evidence to support this hypothesis of heterogeneity was published in 2005 and 2006 by Curtin et al.12,13 These authors divided the cutaneous and mucosal melanomas into the following 4 groups associated with different patterns of sun exposure and anatomical site: 1) melanomas on skin without chronic sun-induced damage (or melanoma related to intermittent sun exposure); 2) melanomas on skin with chronic sun-induced damage (corresponding essentially to melanoma on lentigo maligna); 3) acral melanomas; and 4) mucosal melanomas. They found that while 81% of melanomas on skin without chronic sun-induced damage had BRAF or NRAS mutations (which were mutually exclusive), most melanomas in the other 3 groups, that is, those with a lentiginous histologic pattern, did not have such mutations but CDK4 and CCND1 genes were amplified and/or c-kit genetic aberrations, including mutations and amplifications, were present.
These data have subsequently been extended to provide information on both the types of mutations or abnormalities that are most often detected in the aforementioned genes and the extent to which they can be combined among themselves or with other molecular disorders (for example, loss of PTEN, AKT amplification, etc.) to form different subtypes of melanoma. Thus, therapy using selective targeted drugs in melanoma should take into account this variability and be tailored according to the genetic profile and expression of each tumor subgroup.9,11,14,15
Bcl-2 Antisense Therapy: Other Potential Molecular Targets Implicated in Apoptosis in MelanomaOne of the first directed therapeutic strategies to be tested in clinical trials for the treatment of metastatic melanoma targeted Bcl-2, an antiapoptotic protein that is overexpressed, thereby favoring cell survival. This overexpression occurs in many neoplasms and in 80% of melanomas. One drug, oblimersen can be used as Bcl-2 antisense therapy. The first phase I-II clinical trial in which oblimersen and dacarbazine were combined obtained acceptable results, encouraging design of another trial. In that phase III trial patients were randomized to a group given only dacarbazine or a group given dacarbazine and oblimersen. In total, 771 patients were included. The group that received dacarbazine and oblimersen had a higher objective response rate (13.5% vs 7.5% in the dacarbazine-only group; P=.007) and a longer disease-free interval (median, 2.6 months vs 1.6 months; P<.001). However, there was only a numerical trend towards longer overall survival (median, 9.0 months vs 7.8 months; P=.077). Although a retrospective analysis indicated that the subgroup of patients without elevated serum lactate dehydrogenase hormone (LDH) did benefit in terms of overall survival (median, 11.4 vs 9.7 months; P=.02),16 this finding was not confirmed in subsequent trials. Oblimersen has been combined with other cytostatic agents such as paclitaxel and temozolomide, but the survival outcomes have not improved and so this line of investigation has been abandoned as a possible treatment for melanoma.8,17 Currently, treatments such as ABT-737 and ABT-263 (BH3 mimetics) and YM155 (survivin inhibitor) targeting other antiapoptotic proteins are being tested in clinical trials.17
BRAF InhibitorsRAF is a family of proteins (ARAF, BRAF, and CRAF) that intervene in intracellular signaling of MAP kinases, thereby regulating cell proliferation, differentiation, and survival. BRAF mutations are present in 30% to 70% of melanomas and, as mentioned above, particularly in those associated with chronic sun-induced damage,12,13 the most common type of melanoma. In more than 90% of cases, the BRAF mutation is always the same, a point mutation at exon 15 (thymine replaced by adenine). The result is that valine is replaced by glutamic acid at position 600 of the BRAF protein (V600E mutation). In addition, 15% to 30% of melanomas have NRAS mutations. As NRAS encodes a protein located early in the MAP kinase pathways (Fig. 1), it also acts as a BRAF activator. Interestingly, BRAF mutations have been detected in 20% and NRAS mutations in 80% of common acquired and congenital melanocytic nevi, although the biological significance of this finding is not clear.10 The above findings suggest that BRAF is a very important protein for proliferation of melanocytic cells.18–20
Studies have attempted to find a link between the presence of BRAF and NRAS mutations and the clinical and histopathologic characteristics of melanomas with these mutations.21–23 Although further studies are necessary, in summary, we can now say that melanomas with BRAF mutations are usually more pigmented lesions and are located on the trunk and limbs of middle-aged adults (<50 years) who have a history of sun exposure in childhood, few freckles, and no actinic keratosis. From the histologic point of view, they are found on skin with little actinic elastosis and are more frequently associated with the superficial-spreading variant of melanoma; neoplastic cells tend to invade the upper strata of the epidermis, where they form intraepidermal nests (pagetoid growth pattern); the epidermis is usually thicker and the lesion sharply defined with respect to surrounding skin; the cells are usually rounded, large, and clearly pigmented; and the Breslow depth and mitotic rate are usually lower. Clinically, metastasis tends to be to regional lymph nodes and survival is longer.
SorafenibThe first BRAF inhibitor to be given to patients was BAY43-9006, also known as sorafenib. This drug inhibits the tyrosine-kinase activity of CRAF, but it was soon found to inhibit both wildtype BRAF and the mutated protein (V600E and other mutations) as a well. Subsequently, sorafenib was also found to be a multikinase inhibitor, able to suppress many other targets such as VEGFR2 and VEGFR3, PDGFR-β, p38 MAPK, fms-like tyrosine kinase receptor-3, c-kit, and RET.24 In principle, these actions of sorafenib should not necessarily be negative as melanoma cells have several activated tyrosine-kinase receptors and the VEGFR2 and VEGFR3 activity may even have an antiangiogenic effect. However, although preclinical in vitro studies and testing in animal models were encouraging, the results of clinical trials, which are discussed below, have not confirmed the efficacy of sorafenib for treatment of metastatic melanoma.18–20
In the first phase II trial, which included 39 patients and used sorafenib in monotherapy, 1 complete response and 7 partial responses were achieved. In a second phase I/II trial with 35 patients, when the drug was added to carboplatin and paclitaxel, the number of partial responses increased to 11, while 19 achieved minor responses. Subsequently, a 2-arm phase II trial was undertaken (sorafenib plus dacarbazine vs placebo plus dacarbazine) in which an increase in disease-free interval was observed in the group that received sorafenib, but there was no improvement in overall survival. Other phase III trials have since been undertaken in which sorafenib has been combined with other cytostatic agents (such as temozolomide, carboplatin, or paclitaxel) without achieving any improvement in outcomes. In these trials, response was not correlated with the presence of the BRAF V600E mutation. It is thought that in patients, sorafenib actually targets VEGFR2 or PDGFR-β more strongly than BRAF. In fact, currently, its main indications are treatment of clear cell renal carcinoma and unresectable hepatocellular carcinoma, where angiogenesis seems to play a more important part.18–20 Some preclinical studies suggest that sorafenib would be more effective in a small group of melanomas with BRAF mutations other than the V600E mutation.25
Selective BRAF Inhibitors: PLX4032 and Other AgentsAfter the failure of sorafenib in melanoma, more specific BRAF inhibitors that target the protein encoded by the V600E mutation have been synthesized. The first of these drugs to enter clinical development is PLX4032, which has a low molecular weight and can be administered orally.
Eighty-seven patients were included in the first clinical trial published recently.26 The trial was conducted in 2 phases. In the phase I part, 55 patients were enrolled, 49 with metastatic melanoma (with and without BRAF mutations) and 6 with other neoplasms that usually have BRAF mutations (3 with papillary thyroid carcinoma with the BRAF V600E mutation). A phase II extension enrolled 32 patients, all with melanomas bearing the BRAF V600E mutation. These patients received the dose of PLX4032 established as optimal in the first phase.
Among the 55 patients in the first phase who received a dose of PLX4032≥240mg/12h, 16 had metastatic melanoma with the BRAF V600E mutation. The overall response rate in these patients was 69%. One patient had a complete response. None of the patients with metastatic melanoma without the BRAF V600E mutation had a response even though they had received a dose of PLX4032≥240mg/12h. In the 3 patients with papillary thyroid carcinoma, objective responses were obtained; one of these patients maintained a disease-free interval of 12 months and the other 2 had stable disease for 11 and 13 months.
All 32 patients included in the extension phase had metastatic melanoma with the BRAF V600E mutation and received a 960mg/12h dose of PLX4032. Objective response was observed in 81% of the patients (26/32), with 2 complete responses and 24 partial responses. As observed in the first phase, responses were achieved in patients with visceral metastases to sites usually refractory to treatment (such as metastases to the liver, intestines, and bones), patients with a high serum LDH concentration, and patients who had not responded to other therapies. The median duration of the disease-free period was 7 months. The most noteworthy adverse drug reaction, especially from the dermatological point of view, was the appearance of well-differentiated cutaneous squamous cell carcinomas/keratoacanthomas in 32% of the patients.
Overall, these were the best outcomes ever reported in the treatment of metastatic melanoma. However, given the insufficient follow-up time, the impact of these striking responses on overall survival is still not known. In fact, even though a good response may be achieved, recurrences are generally observed early, usually in a period of 8 to 12 months after treatment.47 These data suggest that resistance to PLX4032 develops easily.28 Initial studies suggest that the mechanisms of resistance to PLX4032 involve reactivation of the MAP kinase pathways not through new BRAF mutations but through development of NRAS mutations, PDGFR-β activation, or overexpression of Cot/tpl2.27,29,30 To prevent resistance from developing, it has been proposed to use PLX4032 in combination with drugs that inhibit other targets such as another molecule implicated in the MAP kinase pathways, for example MEK or the aforementioned COT (see section on Combination Treatments).
The possibility of specifically inhibiting the BRAF V600E mutation has also helped extend our knowledge of interactions between BRAF, CRAF, and activation of the MAP kinase pathways. It has been found that in melanomas without the BRAF V600E mutation (for example, melanomas with the NRAS mutation but with normal or wildtype BRAF), specific inhibition of BRAF induces the formation of BRAF/CRAF dimers which, paradoxically, can activate the MAP kinase pathway and therefore stimulate cell growth. It is therefore very important to use these agents only in those patients with melanomas that bear the V600E mutation.31–35 This mechanism could also explain the onset of cutaneous squamous cell carcinomas/keratoacanthomas, one of the adverse effects of treatment with PLX4032, as proliferation of normal cutaneous epithelial cells, which logically do not have BRAF mutations, is induced.36
Other specific BRAF inhibitors, some of which are now in clinical development, are GSK2118436 (which selectively inhibits the tyrosine-kinase activity of RAF proteins, with a greater potency for BRAF than CRAF), RAF-265 (which intensely inhibits all isoforms of RAF, ARAF, BRAF, and CRAF [including the BRAF V600E mutation gene product], as well as VEGRF-2, c-kit, and PDGFR-β), and XL281 (which acts on the different RAF kinases).28
c-kit Inhibitors: Imatinib (STI-571) and Other Inhibitors of the Tyrosine-Kinase Activity of c-kitc-kit has been considered a potential therapeutic target in melanoma for a long time. In fact, c-kit is a protein that acts as a fundamental growth factor receptor in epidermal melanocytes and has an essential role in the differentiation and migration of melanocytic cells during embryonic development.37 Consistent with this, many melanomas express c-kit. However, in recent years, certain clinical and nonclinical findings have suggested that c-kit is not a valid target. On the one hand, lower c-kit expression as many melanomas progress has been demonstrated,38,39 and we know that in certain cell lines, loss of this growth factor is, paradoxically, related to increased metastatic potential.40,41 On the other, the first clinical trials in patients with metastatic melanoma using c-kit inhibitors, such as imatinib, had discouraging results.42,43
Although there is experimental and clinical evidence of isolated responses to imatinib and of rare mutations in exon 11 of c-kit in some melanoma cells,44–48 this gene was not considered a possible therapeutic target until 2006, when Curtin et al,13 who investigated certain types of uncommon melanomas, established that 39% of mucosal melanomas, 36% of acral melanomas, and 28% of melanomas had c-kit abberations.13 Such aberrations have not been found in uveal melanoma. The low proportion of these types of melanomas in the aforementioned clinical studies of metastatic melanoma would explain the lack of response observed.49 In a clinical trial, designed before these findings were known, but published in 2008, the only patient with response to imatinib had acral lentiginous melanoma.50Genetic aberrations in c-kit include mutations and amplifications of this gene. This point is important because the 2 types of aberration do not necessarily have to have the same biological significance. Of the 39% to 28% of all genetic aberrations of c-kit, only 15% to 38% of mucosal melanomas, 8% to 23% of acral melanomas, and 0 to 17% of melanomas on skin with chronic sun-induced damage are related to mutations. Most mutations, as in gastrointestinal stromal tumors (GIST) in which c-kit mutations are typical, are located on exon 11. However, higher proportions of tumors are associated with mutations located on exons 13, 17, and 18 in melanoma than in GIST and these mutations are in fact the ones associated with resistance to imatinib. Moreover, the coexistence of mutations and amplifications of c-kit has been reported in melanoma but is uncommon in GIST. Studies performed to date seem to indicate that there is not always a relationship between positive and negative immunohistochemical findings for c-kit gene products and the presence or absence of genetic aberrations. Thus, molecular studies seem to be essential to establish whether aberrations of this gene are present.14,51–54
Drugs other than imatinib (sunitinib, dasatinib, and nilotinib) that inhibit the tyrosine-kinase activity of the c-kit gene product are already on the market.3 The ability of these drugs to induce the regression of melanomas with c-kit activating mutations has been shown in several isolated clinical trials2,55–58 and in small series of patients.51 Currently, several phase II studies are in progress with different c-kit inhibitors in patients with metastatic melanoma and this genetic aberration. These trials aim to assess the following aspects: 1) whether the different susceptibility and resistance of various c-kit mutations to the different c-kit inhibitors has significant clinical implications, such that the drug to be used should be selected according to the mutation present in each case; 2) which inhibitors act effectively on metastases of the central nervous system; 3) whether clinical response varies according to whether the melanoma has a c-kit activating mutation or only amplifications, or a combination of an activating mutation and an amplification (in a study of imatinib, a response rate of 50% was attained in patients with melanomas with c-kit mutations, but no responses were observed when only amplification was present59); and 4) how resistance to c-kit inhibitors develops. We know that in GIST, this usually occurs through the appearance of additional c-kit mutations, but it is still not known what the mechanism might be in melanoma.8,52,53
RAS InhibitorsThe isoforms of the RAS oncogene are KRAS, HRAS, and NRAS. Between 15% and 30% of melanomas have NRAS mutations. RAS activating mutations stimulate the MAP kinase pathway and also the PI3K/AKT pathway (Fig. 1) among others. The first drugs used to try to inhibit the MAP kinase pathway were inhibitors of farnesyl transferase of RAS (tipifarnib or R115777). The only phase II study (single-arm design) was terminated due to lack of response. However, none of the patients were selected for the presence of NRAS mutations. There is some evidence that RAS antagonists can enhance the effect of chemotherapy, but this approach has not been tested in clinical trials. We need to see more work on the synthesis of new more effective RAS inhibitors.8 Another alternative would be a concerted inhibition of the 2 main pathways activated as a result of RAS activation, namely, the MAP kinase pathway and the PI3K/AKT pathway14,60 (see section on Combination Treatments).
MEK InhibitorsMEK is a protein that belongs to the MAP kinase pathway downstream from BRAF. MEK inhibitors synthesized to date are PD0325901, AZD6244, GSK1120212, and E6201. The results of the first phase I and phase II trials suggest that these pharmacologic agents are not effective monotherapies in the treatment of melanoma. However, extensive preclinical data suggest that they would be a good choice to use in combined regimens, both for avoiding resistance when using drugs targeting the BRAF V600E mutation and for treating melanoma with BRAF mutations other than V600E or NRAS mutations, especially if combined with inhibitors of the PI3K/AKT pathway8,14,54,61 (see section on Combination Treatments).
PI3K/AKT InhibitorsThe PI3K/AKT pathway is triggered after RAS activation. In melanoma, the pathway is usually highly activated by genetic aberrations that block the physiological regulatory effect of PTEN (NRAS oncogenic mutations [15% to 30% of melanomas], mutations [10% to 20% of melanomas], or silencing of the PTEN tumor suppressor gene) or by AKT amplifications, and recently an uncommon point mutation of AKT3 E17K that activates AKT was identified (Fig. 1).8,10,14,54,61 Various derivatives of rapamycin (CCI-779 or temsirolimus) have been used as inhibitors of the PI3K/AKT pathway. These agents act on mTOR, a molecule located downstream of AKT/PKB (Fig. 1). There are also dual inhibitors of PI3K and mTOR and of PI3K and AKT.62 Although the clinical outcomes in phase-II trials of these drugs have not been good, several authors have proposed they be used in combination therapy, particularly with inhibitors of the MAP kinase pathway8,10,14,54,61 or even in a combination that simultaneously inhibits 2 points on the PI3K/AKT pathway63 (see section on Combination Treatments). However, the results of a study in a murine model of melanoma suggest that inhibition of the PI3K/AKT pathway could induce immunosuppression of the host, thereby favoring tumor growth.64
Inhibition with drugs that target other molecules in other pathways related to proliferation/survival, such as CDK4 or cyclin D1, which are often amplified in melanoma, has not been adequately achieved.60
Inhibitors with Pleiotropic ActionThe term pleiotropic refers to molecules that act on multiple different cell functions. Within this group, we will discuss the proteasome inhibitors and the histone deacetylase inhibitors. These inhibitors affect functions that are important for tumor development and progression, favoring arrest of the cell cycle, apoptosis, decreased invasive and cell migration capacity, generation of reactive oxygen species, inhibition of angiogenesis, and autophagy.
Proteasome InhibitorsThe proteosome is a complex enzyme responsible for the degradation of proteins of intracellular origin. These include proteins implicated in different fundamental cell functions. It has been found that proteasome inhibition induces cell apoptosis, both in normal cells and in malignant cells, although the effect is stronger in the latter case. In addition, proteasome inhibition indirectly enhances cell death induced by other proapoptotic mechanisms such as chemotherapy or radiotherapy.65 Proteasome inhibitors, and bortezomib (PS-341) in particular, are used in the treatment of multiple myeloma and other hematologic malignancies. However, they do not appear to be as effective in solid tumors. These inhibitors have been shown to be able to induce apoptosis and inhibit cell growth in both in vitro and in vivo experimental models of melanoma.66–68 Nevertheless, satisfactory outcomes were not obtained in the first clinical trial in metastatic melanoma, in which bortezomib was used in monotherapy.69 The findings of a phase I trial that combined bortezomib with temozolomide were likewise not very encouraging.70 Nevertheless, the possibility of treatment combined with other therapies continues to be explored71–79 and we are now seeing a new generation of proteasome inhibitors in development.80
Histone Deacetylase InhibitorsOne of the characteristics of neoplastic cells is aberrant regulation of gene expression modulated by epigenetic mechanisms. These heritable mechanisms can amplify the variability in genome expression in an individual by regulating the expression of that person's genes without actually changing any DNA sequences. One of the best-known epigenetic mechanisms is gene silencing by methylation of the promoter. Histone acetylation is another such mechanism. Histone deacetylases are a group of enzymes that silence genes by eliminating acetyl groups from the histones. As these enzymes act on key genes in the cell cycle, their inhibition leads to multiple cell alterations. The histone deacetylase inhibitor known as vorinostat (suberoylanilide hydroxamic acid) has already been approved for treatment of T-cell cutaneous lymphomas.81,82 In melanoma, encouraging results have been obtained with the use of several histone deacetylase inhibitors in in vitro or animal models,83–87 especially in combination with other treatments such as cytostatic agents, radiotherapy, retinoids, immunotherapy, or other targeted agents.88–92 Nevertheless, the results of the first phase I and phase II clinical trials, whether they were testing the inhibitors in monotherapy or combination therapy, have shown only limited efficacy.93–96
Molecular Targets Implicated in Invasive and Metastatic Mechanisms: Antiadhesion Molecule TherapyThe high invasive and migratory capacity of melanoma cells can be partly attributed to expression of an anomalous pattern of adhesion molecules, a characteristic that differentiates them from normal melanocytic cells. For example, this capacity helps the melanoma cells detach themselves from epidermal keratinocytes (loss of caderin E), adhere to fibroblasts and the vascular endothelium (overexpression of caderin N), switch from the radial growth phase to vertical growth and metastasize (overexpression of melanoma cell adhesion molecule [MCAM]) or adhere to proteins of the extracellular matrix and secrete metalloproteinases to degrade these proteins (overexpression of integrin αvβ3).97,98 In addition, the inhibition of some types of adhesion molecules, such as integrins, may also inhibit proliferation and induce apoptosis in neoplastic cells as well as exercise an antiangiogenic effect.99,100
One of the main therapeutic targets is integrin αvβ3, against which there are monoclonal antibodies such as etaracizumab (MEDI-522), whose antitumor activity as a single agent or in combination with MCAM, or caderin N inhibitors was demonstrated in preclinical studies.101 However, on phase I and phase II clinical testing, both in monotherapy or in combination with cytostatic agents, the results were not encouraging.102 Caderin N inhibitor (ADH-1) therapy has also proven disappointing in trials.103 Overall, therapy targeting adhesion molecules does not seem, for the time being, to be very promising for the treatment of metastatic melanoma.97,98
Therapeutic Targets in Structures Other Than Neoplastic CellsTherapeutic Targets in the Tumor Environment: Antiangiogenic DrugsMelanoma is a tumor associated with intense angiogenesis, which allows and sustains tumor growth and facilitates metastatic activity. Antiangiogenic therapy may therefore be another useful therapeutic strategy in melanoma.104 One of the most widely tested antiangiogenic drugs in clinical trials is bevacizumab, a monoclonal antibody that targets VEGF. In melanoma it has been used as a single agent, without much success, or in combination with cytostatic agents (carboplatin plus paclitaxel or temozolomide) or with interferon alfa. In one of the most recent phase II studies presented in the conference of the European Cancer Organisation-European Society for Medical Oncology in 2009, 214 patients received either carboplatin, paclitaxel, and bevacizumab or carboplatin, paclitaxel, and placebo.105 The group that received bevacizumab tended to have a higher response rate as well as higher overall survival and 1-year survival rates, but none of these differences were statistically significant.8,17,61,100
Other proposed antiangiogenic drugs, although ones so far without any noteworthy clinical effects, are multikinase inhibitors of the different VEGF receptors (VEGFR-1, 2, and -3)—sunitinib, sorafenib, semaxinib, axatinib etc.—thalidomide derivatives, and antiadhesion molecules.8,17,61,100,104,106
Therapeutic Targets That Enhance Tumor Escape Mechanisms: Anti-CTLA-4 Antibodies, and Other Types of Immunotherapy Directed Against Molecular TargetsAlthough melanoma is an extremely immunogenic tumor that readily triggers an immune response against tumor cells, these use a series of evasion strategies. One way of fighting against these escape mechanisms is to inhibit a T-lymphocyte receptor, namely cytotoxic T-lymphocyte antigen (CTLA) 4, which induces immune tolerance. CTLA-4 competes with the CD28 on lymphocytes in binding to the B7 molecule of antigen presenting cells. CTLA-4/B7 binding induces anergy instead of stimulating the cytotoxic immune response. Thus, inhibition of CTLA-4 could overcome immune tolerance to melanoma. Two anti-CTLA-4 monoclonal antibodies have been used in patients with metastatic melanoma, ipilimumab and tremelimumab.107 Ipilimumab is the one that has been developed more quickly and with greater clinical success.107–109 Indeed, ipilimumab has recently been approved by the US Federal Drug Administration for the treatment of metastatic melanoma.
Initial assessment of clinical responses to this type of drug has been problematic. The problem that arises when evaluating response to immunomodulatory agents, such as anti-CTLA-4 antibodies, is that they can induce a biphasic response characterized by an initial worsening followed by subsequent long-term benefit. This biphasic pattern of response cannot be appropriately assessed with the systems currently used in oncology (RECIST [Response Evaluation Criteria In Solid Tumors] or the World Health Organization criteria). Thus, new criteria have been drawn up to specifically evaluate this type of response.107,108 These criteria place higher value on overall survival and stabilization of the disease than on the initial response assessed by traditional methods.
The most recent phase III trial of ipilimumab included 676 patients randomized to 3 different arms.109 One arm (with 403 patients) received ipilimumab along with the gp100 peptide vaccine, another arm (with 177 patients) received only ipilimumab, and the third arm received the vaccination only. No significant differences were observed between the 2 arms that received ipilimumab. However, the median overall survival in patients who received ipilimumab and gp100 vaccine was significantly longer than that in patients who received the vaccine only (10 months vs 6.4 months, respectively; P<.001). Taken together, the findings of the clinical trials published to date indicate that, as with BRAF inhibitors, one of the advantages of ipilimumab is that it induces responses in patients with metastasis with poor prognosis, including patients with metastasis to the central nervous system and/or those with high serum LDH levels. Another of the inherent advantages of this type of treatment is that once response is achieved (10%, complete or partial response; 10% to 20%, disease stabilization; and 10%, initial disease progression but subsequent clinical response), the response is sustained, with follow-up of up to 5 years in some cases. There is also the possibility of re-treating patients, in the event of relapse, months or years after the end of the initial treatment.107 The main side effects are those related to the immune system—dermatitis, colitis, hypophysitis, thyroiditis, and hepatitis—and these reach grade 3 or more in 10% to 15%. Colon perforation may occur, and so patients with diarrhea should be closely monitored. Specific algorithms have been developed to allow safer management of the toxicities specific to anti-CTLA-4 antibodies. One line of investigation receiving much attention is the search for predictive markers to select patients who may benefit from treatment. Appropriate selection is important, in terms of deciding to persist with treatment when there is initial disease progression as well as avoiding toxicity, which can be severe, in patients who are potentially nonresponders.107,110
Combination therapy with drugs aimed at other targets with cytostatic agents is also being tested and the role of these new agents in high-risk melanoma is being assessed.107,110 Other targets that could be inhibited with a similar strategy and for which studies are being conducted are CD137, OX40, and PD1.2,110
Combination TreatmentsFrom the clinical outcomes obtained so far, it seems unlikely that stable disease remission could be achieved by working with a single therapeutic target. An explanation for the onset of resistance to the treatments that have been tried is that melanoma cells develop compensatory responses. Alternatively, redundant signaling pathways might be activated when other pathways are interrupted. Therefore, a strategy for avoiding or combating these processes would be the simultaneous use of drugs directed against more than one target. Two combinations enjoy the most support. One is the simultaneous inhibition of 2 targets in the MAP kinase pathway (especially BRAF and MEK in what could be termed vertical inhibition of this pathway). Another is inhibition of a target in the MAP kinase pathway and a target in the PI3K/AKT pathway. Recently, the vertical inhibition of the PI3K/AKT pathway, by acting simultaneously on PI3K and mTOR, has been proposed.63 However, there are many other possible combinations with conventional cytostatic agents, molecules with pleiotropic action, different forms of immunotherapy, and any treatment aimed at the targets discussed above.8,54,60,97,110,112
In addition to the combinations already mentioned, the list of possible targets under investigation for the treatment of melanoma is very long and will surely continue to grow. On the list at present are Hsp90, ERBB4, GNaQ, SSTRs, CDK2, iNOS, FGFR-1, APP, and more.14,54,97,111 Of all the drugs tested to date, the most promising overall are specific BRAF inhibitors in melanomas with the BRAF V600E mutation, c-kit inhibitors in cutaneous and mucosal lentiginous melanomas with c-kit mutations, and the anti-CTLA-4 monoclonal antibody, able to suppress the development of immune tolerance in melanoma. Although these options are a significant improvement compared to those available a few years ago, there are still many problems to resolve. First, it is important to design strategies to avoid the onset of resistance to BRAF and c-kit inhibitors. If this is not possible, strategies should be available to manage this resistance. It is also essential to identify biomarkers that allow the selection of patients potentially susceptible to therapies such as ipilimumab. Targets should also be sought for effective treatment in patients with mutations other than BRAF V600E or c-kit mutations. Finally, the feasible options of combination therapy should be studied. These problems can best be overcome if full advantage is taken of all the information generated by clinical trials. These trials, for their part, should obviously be designed so that their findings can be used (for example, they should enroll patients with suitably defined characteristics and collect samples during the trial for a variety of molecular studies). Preclinical research should also continue. For the first time, there is the feeling that a new horizon has opened up in the treatment of metastatic melanoma. However, we have only taken the first few steps; the real journey has hardly begun.20,60
FundingThis article was supported by grants FIS-PI060832, 2009SGR794, and RD06/0020/1034, a predoctoral grant from the Asociación Española Contra el Cáncer, Catalunya contra el Càncer, Lleida, Spain, for A.S. and a postdoctoral contract Juan de la Cierva for A.Y.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Martí RM, Sorolla A y Yeramian A. Nuevas dianas terapéuticas en el melanoma. Actas Dermosifiliogr.2012;103:579-590.



